As filed with the United States Securities and Exchange Commission on February 1, 2021.
Registration Statement No. 333-252166
England and Wales | | | 2836 | | | Not applicable |
(State or other jurisdiction of incorporation or organization) | | | (Primary Standard Industrial Classification Code Number) | | | (I.R.S. Employer Identification Number) |
Divakar Gupta Eric W. Blanchard Peter Byrne Courtney T. Thorne Cooley LLP 55 Hudson Yards New York, New York 10001 +1 212 479 6000 | | | Nicola Maguire Claire Keast-Butler Thomas Goodman Cooley (UK) LLP Dashwood 69 Old Broad Street London EC2M 1QS United Kingdom +44 20 7583 4055 | | | Simon Witty Davis Polk & Wardwell London LLP 5 Aldermanbury Square London EC2V 7HR United Kingdom +44 20 7418 1300 | | | Richard D. Truesdell, Jr. Yasin Keshvargar Davis Polk & Wardwell LLP 450 Lexington Avenue New York, New York 10017 +1 212 450 4000 |
Title of Each Class of Securities to be Registered | | | Amount to be Registered(1) | | | Proposed Maximum Offering Price Per Share(2) | | | Proposed Maximum Aggregate Offering Price(1)(2) | | | Amount of Registration Fee(5) |
Ordinary shares, nominal value £0.002 per share(3)(4) | | | 9,583,332 | | | $25.00 | | | $239,583,300 | | | $26,139 |
(1) | Includes 1,249,999 shares that the underwriters have the option to purchase. |
(2) | Estimated solely for the purpose of calculating the registration fee in accordance with Rule 457(a) of the Securities Act of 1933, as amended. |
(3) | These ordinary shares are represented by ADSs, each of which represents one ordinary share of the Registrant. |
(4) | ADSs issuable upon deposit of the ordinary shares registered hereby are being registered pursuant to a separate registration statement on Form F-6 (File No. 333-252487). |
(5) | The registrant previously paid a registration fee of $10,910 in connection with the initial filing of this Registration Statement. |
1 | We have submitted an application to the Registrar of Companies in England and Wales for re-registration as a public limited company with the name Immunocore Holdings plc. |
† | The term “new or revised financial accounting standards” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012. |
| | | PER ADS | | | TOTAL | |
Initial public offering price | | | $ | | | $ |
Underwriting discounts and commissions(1) | | | | | ||
Proceeds, before expenses, to us | | | | |
(1) | See “Underwriting” for additional information regarding total underwriter compensation. |
Goldman Sachs & Co. LLC | | | J.P. Morgan | | | Jefferies |

• | Tebentafusp, our ImmTAC molecule targeting an HLA-A*02:01 gp100 antigen, demonstrated monotherapy activity and recently achieved the primary endpoint of superior overall survival at the first pre-planned interim analysis of a randomized Phase 3 clinical trial in patients with previously untreated metastatic uveal melanoma. We anticipate submitting a BLA to the FDA in the third quarter of 2021 followed by a Marketing Authorization Application, or MAA, submission to the European Medicines Agency, or EMA. |
• | IMC-C103C, our ImmTAC molecule targeting an HLA-A*02:01 MAGE-A4 antigen, is currently being evaluated in a first-in-human, Phase 1/2 dose escalation trial in patients with solid tumor cancers including non-small-cell lung cancer, or NSCLC, gastric, head and neck, ovarian and synovial sarcoma. We believe this trial will demonstrate clinical activity of IMC-C103C, and we anticipate reporting Phase 1 initial data from this trial in the second half of 2021. We are developing this program under a co-development collaboration with Genentech, Inc., or Genentech, under which we have an option to retain 50% of the economics. |
• | IMC-F106C, our ImmTAC molecule targeting an optimal HLA-A*02:01 PRAME antigen identified with our MassSpec technology, is currently being evaluated in a first-in-human, Phase 1/2 dose escalation trial in patients with multiple solid tumor cancers including breast, endometrial, ovarian and small cell lung cancer, or SCLC. We believe this trial will demonstrate clinical activity of IMC-F106C, and we anticipate reporting Phase 1 initial data from this trial in mid-2022. |
• | GSK01, our ImmTAC molecule targeting an NY-ESO HLA-A*02:01 antigen, is currently being evaluated in the dose escalation phase of a Phase 1 clinical trial. When an optimal dosing regimen has been identified, a small expansion cohort of synovial sarcoma patients will be recruited to evaluate the clinical benefit of the therapeutic. This program is being developed under a collaboration with GlaxoSmithKline Intellectual Property Development Ltd, or GSK, which has an option to acquire full commercialization and development rights to this product candidate at the end of the ongoing Phase 1 clinical trial. |
• | IMC-I109V, our ImmTAV molecule targeting a conserved HBV envelope antigen, is our most advanced ImmTAV program and is currently being evaluated in a Phase 1/2 clinical trial in patients with chronic HBV who are non-cirrhotic, hepatitis B e-Antigen negative, and virally suppressed on chronic nucleot(s)ide analogue therapy. Our goal is to develop a functional cure for HBV and we anticipate commencing dosing in our Phase 1 single ascending dose, or SAD, trial in mid-2021. We are also developing a next-generation version of this molecule leveraging our research into universal HLA-E molecules which could benefit a much larger patient population as compared to classical-HLA antigens. |
• | IMC-M113V, our ImmTAV molecule targeting an HIV gag antigen bispecific TCR molecule, is currently in pre-clinical development. Our HIV programs are funded by the Bill & Melinda Gates Foundation, or the Gates Foundation, and we are required to make any successfully approved products available at reduced prices in certain developing countries. We retain full development and commercial right in non-developing countries. |
• | Secure marketing approval for, and then commercialize, tebentafusp, our lead ImmTAC, for the treatment of metastatic uveal melanoma. |
• | Advance our IMC-C103C program targeting MAGE-A4 for the treatment of solid tumors in collaboration with Genentech. |
• | Advance our IMC-F106C program targeting PRAME for the treatment of solid tumors. |
• | Advance our IMC-I109V program for the treatment of chronic HBV. |
• | Continue to develop our novel universal ImmTAX platform to meaningfully broaden the eligible patient pool. |
• | Continue to invest in our platform to discover and develop novel therapeutics. |
• | Opportunistically pursue strategic partnerships to maximize the full potential of our pipeline and ImmTAX platform. |
• | We have incurred significant losses in every year since our inception. We expect to continue to incur losses over the next several years and may never achieve or maintain profitability. |
• | We will require substantial additional funding to achieve our business goals. If we are unable to obtain this funding when needed and on acceptable terms, we could be forced to delay, limit or terminate our product development efforts. |
• | We are heavily dependent on the success of our ImmTAX platform to identify and develop product candidates. If we or our collaborators are unable to successfully develop and commercialize our platforms or experience significant delays in doing so, our business may be harmed. |
• | We may be unable to successfully complete additional large-scale, pivotal clinical trials for any product candidates we develop after tebentafusp. |
• | Our product candidates utilize a novel mechanism of action and involve novel targets which may result in greater research and development expenses, regulatory issues that could delay or prevent approval, or discovery of unknown or unanticipated adverse effects. |
• | Clinical product development involves a lengthy and expensive process, with an uncertain outcome. |
• | The effects of health epidemics, including the ongoing COVID-19 coronavirus pandemic, in regions where we, or the third parties on which we rely, have business operations could adversely impact our business, including our pre-clinical studies and clinical trials, as well as the business or operations of our CROs or other third parties with whom we conduct business. |
• | For a period of four weeks, our IMC-F106C program was put on partial clinical hold in 2020 by the FDA following the death of the second patient dosed in this trial, which was subsequently determined to be unrelated to study drug. The hold has since been lifted and the trial has been resumed. |
• | We are subject to manufacturing risks that could substantially increase our costs and limit supply of our products. |
• | We face substantial competition, which may result in others developing or commercializing drugs before or more successfully than us. |
• | Our existing collaborations are important to our business, and future collaborations may also be important to us. If we are unable to maintain any of these collaborations, or if these collaborations are not successful, our business could be adversely affected. |
• | If we are unable to adequately protect our proprietary technology or obtain, maintain, protect and enforce patent and other intellectual property protection for our technology and products or if the scope of the protection obtained is not sufficiently broad, our competitors and other third parties could develop and commercialize technology and products similar or identical to ours, and our ability to successfully commercialize our technology and products may be impaired. |
• | Third parties may initiate legal proceedings alleging that we are infringing, misappropriating or otherwise violating their intellectual property or proprietary rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business. |
• | The FDA regulatory pathways can be difficult to predict and whether, for example, further unanticipated clinical trials are required, will depend on the data obtained in our ongoing clinical trials. |
• | Our future success depends on our ability to retain key executives and experienced scientists and to attract, retain and motivate qualified personnel. |
• | As a company based outside of the United States, we are subject to economic, political, regulatory and other risks associated with international operations. |
• | We have identified a material weakness in our internal control over financial reporting and may identify material weaknesses in the future or otherwise fail to maintain proper and effective internal controls, which may impair our ability to produce timely and accurate financial statements or prevent fraud. If we are unable to establish and maintain effective internal controls, shareholders could lose confidence in our financial and other public reporting, which would harm our business and the trading price of our ADSs. |
• | an exemption from compliance with any requirement that the Public Company Accounting Oversight Board may adopt regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements; |
• | reduced disclosure about our executive compensation arrangements; |
• | an exemption from the non-binding advisory votes on executive compensation, including golden parachute arrangements; and |
• | an exemption from the auditor attestation requirement in the assessment of our internal control over financial reporting pursuant to the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act. |
• | to fund tebentafusp, our lead ImmTAC, for the treatment of metastatic uveal melanoma through the completion of our Phase 3 clinical trial as well as preparations for a commercial launch; |
• | to advance the clinical development of IMC-C103C targeting MAGE A4 for the treatment of solid tumors; |
• | to advance the clinical development of IMC-F106C targeting PRAME for the treatment of solid tumors; |
• | to advance the clinical development of IMC-I109V targeting a functional cure for chronic HBV; |
• | to continue to continue to advance our pre-clinical programs and invest in our ImmTAX platform to discover and develop novel therapeutic targets; and |
• | for working capital and general corporate purposes. |
• | 832,904 ordinary shares issuable upon the exercise of options outstanding under our existing equity incentive plans as of September 30, 2020, with a weighted-average exercise price of £63.23 per share (or $81.70 per share, based on the noon buying rate of the Federal Reserve Bank of New York on September 30, 2020, which was £1.00 to $1.2921); |
• | 5,706,279 ordinary shares reserved for future issuance under our 2021 Equity Incentive Plan, or the 2021 EIP, which will become effective in connection with this offering, as well as any automatic annual increases in the number of ordinary shares reserved for future issuance under the 2021 EIP, as more fully described in the section titled “Management—Equity Incentive Plans;” and |
• | 4,014,267 ordinary shares (assuming an initial public offering price of $24.00 per ADS, which is the midpoint of the price range set forth on the cover page of this prospectus) underlying the grants to be issued prior to the closing of this offering to certain of our officers, directors and employees under our 2021 EIP, contingent and effective upon the execution and delivery of the underwriting agreement relating to this offering, with an exercise price that is equal to or greater than the price per ADS at which our ADSs are first sold to the public in this offering. |
• | the completion of the transactions described in the section titled “Corporate Reorganization” prior to the completion of this offering, including (i) the 20 to 1 consolidation of all our ordinary shares and non-voting ordinary shares prior to completion of this offering, (ii) the re-designation of G1 shares held by our U.K. employees and former employees as deferred shares; (iii) 216,200 ordinary shares underlying the grants to be issued prior to the closing of this offering under our 2021 EIP (to our current employees) and under standalone option agreements (to our former employees) to replace the G1 shares that will be re-designated as deferred shares, conditional on and effective immediately prior to closing of this offering, with an exercise price that is equal to or greater than the price per ADS at which our ADSs are first sold to the public in this offering; (iv) the re-designation of G2 shares held by our U.K. employees into one ordinary share and three deferred shares, conditional on and effective immediately prior to closing of this offering; and (v) 96,300 ordinary shares underlying the grants to be issued prior to the closing of this offering under our 2021 EIP to replace the G2 shares that will be re-designated as a mixture of deferred shares and ordinary shares, conditional on and effective immediately prior to closing of this offering, with an exercise price that is equal to the price per ADS at which our ADSs are first sold to the public in this offering; |
• | an initial public offering price of $24.00 per ADS, which is the midpoint of the price range set forth on the cover page of this prospectus; and |
• | no exercise by the underwriters of their option to purchase up to 1,249,999 additional ADSs in this offering. |
| | | For the nine month period ended September 30, | | | For the year ended December 31, | |||||||
| | | 2020 | | | 2019 | | | 2019 | | | 2018 | |
| | | (pounds sterling, in thousands, except for share and per share data) | | | (pounds sterling, in thousands, except for share and per share data) | |||||||
Consolidated statement of loss and other comprehensive income data: | | | | | | | | | ||||
Revenue | | | 22,694 | | | 22,027 | | | 25,669 | | | 23,654 |
Other operating income | | | 408 | | | 420 | | | 185 | | | 622 |
Operating expenses: | | | | | | | | | ||||
Research and development | | | (57,566) | | | (75,415) | | | (99,991) | | | (83,575) |
Administrative expenses | | | (31,569) | | | (35,611) | | | (44,183) | | | (34,156) |
Operating loss | | | (66,033) | | | (90,579) | | | (118,320) | | | (93,455) |
Other income | | | — | | | — | | | — | | | 4,979 |
Finance income | | | 1,972 | | | 1,134 | | | 1,510 | | | 1,140 |
Finance costs | | | (2,272) | | | (6,532) | | | (9,379) | | | (842) |
Non-operating (expense) / income | | | (300) | | | (5,398) | | | (7,869) | | | 5,277 |
Loss before tax | | | (66,333) | | | (95,977) | | | (126,189) | | | (88,178) |
Income tax credit | | | 11,120 | | | 18,011 | | | 22,258 | | | 16,548 |
Loss for the period | | | (55,213) | | | (77,966) | | | (103,931) | | | (71,630) |
Exchange differences on translation of foreign operations | | | 338 | | | 82 | | | (99) | | | 72 |
Income tax effect relating to the components of other comprehensive income | | | — | | | — | | | — | | | 3,634 |
Total comprehensive loss for the period, net of tax | | | (54,875) | | | (77,884) | | | (104,030) | | | (67,924) |
| | ||||||||||||
Pro forma per share data:(1) | | | | | | | | | ||||
Basic and diluted loss per share(2) | | | (0.002) | | | (0.004) | | | (0.005) | | | (0.003) |
Basic and diluted weighted average number of shares | | | 27,306,935 | | | 21,579,880 | | | 22,297,935 | | | 21,558,890 |
| | | As of September 30, | | | As of December 31, | ||||
| | | 2020 | | | 2019 | | | 2018 | |
| | | (pounds sterling, in thousands) | | | (pounds sterling, in thousands) | ||||
Consolidated statement of financial position data: | | | | | | | |||
Cash and cash equivalents(3) | | | 56,687 | | | 73,966 | | | 124,385 |
Working capital(4) | | | 29,335 | | | 39,768 | | | 121,574 |
Total assets | | | 130,839 | | | 185,649 | | | 195,777 |
Interest-bearing loans and borrowings(3) | | | — | | | 19,157 | | | 18,878 |
Total liabilities(2) | | | 115,291 | | | 170,878 | | | 139,195 |
Share capital | | | 1 | | | — | | | — |
Total equity | | | 15,548 | | | 14,771 | | | 56,582 |
(1) | The unaudited pro forma loss per share data gives effect to our corporate reorganization. |
(2) | See Note 10 to our audited consolidated financial statements for the year ended December 31, 2019 and year ended December 31, 2018 and Note 6 to our unaudited consolidated financial statements appearing elsewhere in this prospectus for a description of the method used to compute diluted net loss per share. |
(3) | Excludes the borrowing of $50.0 million in November 2020 under a new debt facility with Oxford Finance Luxembourg S.A.R.L. |
(4) | We define working capital as current assets less current liabilities. |
• | continue our ongoing and planned development of our five clinical stage programs, including tebentafusp, our lead oncology program, which is being evaluated in a Phase 3 pivotal trial in patients with metastatic uveal melanoma; |
• | initiate pre-clinical studies and clinical trials for any additional product candidates that we may pursue in the future, including our earlier-stage programs; |
• | seek regulatory approvals for tebentafusp and any future product candidates that successfully complete clinical trials; |
• | build a portfolio of product candidates through the discovery, development, or acquisition or in-license of drugs, product candidates or technologies; |
• | establish a sales, marketing, manufacturing and distribution capability to commercialize tebentafusp and any future product candidate for which we may obtain marketing approval; |
• | maintain, protect, enforce and expand our intellectual property portfolio; |
• | acquire or in-license other product candidates, intellectual property and technologies; |
• | hire additional clinical, regulatory and scientific personnel; |
• | add operational, financial and management information systems and personnel, including personnel to support our product development and planned future commercialization efforts; and |
• | incur additional legal, accounting and other expenses associated with operating as a public company. |
• | progress, timing, scope and costs of our clinical trials, including the ability to timely initiate clinical sites, enroll subjects and manufacture soluble bispecific TCR product candidates for our ongoing, |
• | time and costs required to perform research and development to identify and characterize new product candidates from our research programs; |
• | the time and cost necessary to pursue regulatory authorizations and approvals that may be required by regulatory authorities to execute clinical trials or commercialize our products; |
• | our ability to successfully commercialize our product candidates, if approved; |
• | our ability to have clinical and commercial products successfully manufactured consistent with FDA, EMA and other authorities’ regulations; |
• | amount of sales and other revenues from product candidates that we may commercialize, if any, including the selling prices for such potential products and the availability of adequate third-party coverage and reimbursement for patients; |
• | sales and marketing costs associated with commercializing our products, if approved, including the cost and timing of building our marketing and sales capabilities; |
• | cost of building, staffing and validating our manufacturing processes, which may include capital expenditure; |
• | terms and timing of any revenue from our existing collaborations; |
• | costs of operating as a public company; |
• | time and cost necessary to respond to technological, regulatory, political and market developments; |
• | costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; |
• | costs, associated with, and terms and timing of, any future any potential acquisitions, strategic collaborations, licensing agreements or other arrangements that we may establish; and |
• | inability of clinical sites to enroll patients as healthcare capacities are required to cope with natural disasters, epidemics or other health system emergencies, such as the COVID-19 pandemic. |
• | be delayed in obtaining marketing approval for our product candidates; |
• | not obtain marketing approval at all; |
• | obtain approval for indications or patient populations that are not as broad as intended or desired; |
• | be subject to post-marketing testing requirements; or |
• | have the product removed from the market after obtaining marketing approval. |
• | the research methodology used may not be successful in identifying potential indications and/or product candidates; |
• | potential product candidates may, after further study, be shown to have harmful adverse effects or other characteristics that indicate they are unlikely to be effective products; or |
• | it may take greater human and financial resources than we will possess to identify additional therapeutic opportunities for our product candidates or to develop suitable potential product candidates through internal research programs, thereby limiting our ability to develop, diversify and expand our product portfolio. |
• | delays or difficulties in enrolling and retaining patients in our clinical trials, including patients that may not be able or willing to comply with clinical trial protocols such as weekly dosing regimens if quarantines impede patient movement or interrupt healthcare services; |
• | delays or difficulties in clinical site initiation, including difficulties in recruiting and retaining clinical site investigators and clinical site staff; |
• | increased rates of patients withdrawing from our clinical trials following enrollment as a result of risks of exposure to COVID-19, being forced to quarantine or being unable to visit clinical trial locations or otherwise comply with clinical trial protocols; |
• | diversion or prioritization of healthcare resources away from the conduct of clinical trials and towards the COVID-19 pandemic, including the diversion of hospitals serving as our clinical trial sites and hospital staff supporting the conduct of our clinical trials, and because, who, as healthcare providers, may have heightened exposure to COVID-19 and adversely impact our clinical trial operations; |
• | interruption of our clinical supply chain or key clinical trial activities, such as clinical trial site monitoring, due to limitations on travel imposed or recommended by federal, state/provincial or municipal governments, employers and others; and |
• | limitations in employee resources that would otherwise be focused on the conduct of our clinical trials, including because of sickness of employees or their families or the desire of employees to avoid contact with large groups of people. |
• | delays in receiving approval from local regulatory authorities to initiate our planned clinical trials; |
• | delays in clinical sites receiving the supplies and materials needed to conduct our clinical trials; |
• | interruption in global shipping that may affect the transport of clinical trial materials, such as investigational drug product and comparator drugs used in our clinical trials; |
• | changes in federal, state/provincial or municipal regulations as part of a response to the COVID-19 coronavirus outbreak which may require us to change the ways in which our clinical trials are conducted, which may result in unexpected costs, or to discontinue the clinical trials altogether; |
• | delays in necessary interactions with local regulators, ethics committees and other important agencies and contractors due to limitations in employee resources or forced furlough of government employees; and |
• | the refusal of the FDA to accept data from clinical trials in these affected geographies. |
• | the FDA, EMA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials; |
• | the FDA, EMA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials; |
• | we may be unable to demonstrate to the satisfaction of the FDA, EMA or comparable foreign regulatory authorities that a product candidate is safe and effective for its proposed indication or a related companion diagnostic is suitable to identify appropriate patient populations; |
• | the results of clinical trials may not meet the level of statistical significance required by the FDA, EMA or comparable foreign regulatory authorities for approval; |
• | we may be unable to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks; |
• | the FDA, EMA or comparable foreign regulatory authorities may disagree with our interpretation of data from pre-clinical studies or clinical trials; |
• | the data collected from clinical trials of our product candidates may not be sufficient to support the submission a BLA or other submission or to obtain regulatory approval in the United States or elsewhere; |
• | the FDA, EMA or comparable foreign regulatory authorities may find deficiencies with or fail to approve the manufacturing processes or facilities of third-party manufacturers with which we contract for clinical and commercial supplies; and |
• | the approval policies or regulations of the FDA, EMA or comparable foreign regulatory authorities may significantly change such that our clinical data are insufficient for approval. |
• | regulators or institutional review boards, or IRBs, or ethics committees may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site; |
• | we may experience delays in reaching, or fail to reach, agreement on acceptable terms with prospective trial sites and prospective CROs, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
• | clinical trials of our product candidates may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional pre-clinical studies or clinical trials or we may decide to abandon product development programs; |
• | the number of patients required for clinical trials of our product candidates may be larger than we anticipate, enrollment in these clinical trials may be slower than we anticipate or participants may drop out of these clinical trials or fail to return for post-treatment follow-up at a higher rate than we anticipate; |
• | our third-party contractors may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all, or may deviate from the clinical trial protocol or drop out of the trial, which may require that we add new clinical trial sites or investigators; |
• | we may elect to, or regulators or IRBs or ethics committees may require us or our investigators to, suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements or a finding that the participants are being exposed to unacceptable health risks; |
• | the cost of clinical trials of our product candidates may be greater than we anticipate; |
• | the supply or quality of our product candidates or other materials necessary to conduct clinical trials of our product candidates may be insufficient or inadequate; and |
• | our product candidates may have undesirable side effects or other unexpected characteristics, causing us or our investigators, regulators or IRBs or ethics committees to suspend or terminate the trials, or reports may arise from pre-clinical or clinical testing of other cancer therapies that raise safety or efficacy concerns about our product candidates. |
• | the severity of the disease under investigation; |
• | the eligibility criteria for the clinical trial in question; |
• | the availability of an appropriate genomic screening test; |
• | the perceived risks and benefits of the product candidate under study; |
• | the efforts to facilitate timely enrollment in clinical trials; |
• | the patient referral practices of physicians; |
• | the ability to monitor patients adequately during and after treatment; |
• | the proximity and availability of clinical trial sites for prospective patients; and |
• | factors we may not be able to control, such as current or potential pandemics that may limit patients, principal investigators or staff or clinical site availability (e.g., outbreak of COVID-19). |
• | our available capital resources or capital constraints we experience; |
• | the rate of progress, costs, and results of our clinical trials and research and development activities, including the extent of scheduling conflicts with participating clinicians and collaborators; |
• | our ability to identify and enroll patients who meet clinical trial eligibility criteria; |
• | our receipt of approvals by the FDA, EMA and comparable foreign regulatory authorities, and the timing thereof; |
• | other actions, decisions, or rules issued by regulators; |
• | our ability to access sufficient, reliable, and affordable supplies of materials used in the manufacture of our product candidates; |
• | our ability to manufacture and supply clinical trial materials to our clinical sites on a timely basis; |
• | the efforts of our collaborators with respect to the commercialization of our approved products, if any; and |
• | the securing of, costs related to, and timing issues associated with, commercial product manufacturing, as well as sales and marketing activities. |
• | differing regulatory requirements in foreign countries; |
• | unexpected changes in tariffs, trade barriers, price and exchange controls and other regulatory requirements; |
• | differing standards for the conduct of clinical trials; |
• | increased difficulties in managing the logistics and transportation of storing and shipping product candidates produced in the United States or elsewhere and shipping the product candidate to patients in other countries; |
• | import and export requirements and restrictions; |
• | economic weakness, including inflation, or political instability in foreign economies and markets; |
• | compliance with tax, employment, immigration and labor laws for employees living or traveling abroad; |
• | foreign taxes, including withholding of payroll taxes; |
• | foreign currency fluctuations, which could result in increased operating expenses and reduced revenue, and other obligations incident to doing business in another country; |
• | difficulties staffing and managing foreign operations; |
• | workforce uncertainty in countries where labor unrest is more common than in the United Kingdom or the United States; |
• | differing payor reimbursement regimes, governmental payors or patient self-pay systems, and price controls; |
• | potential liability under the Foreign Corrupt Practices Act of 1977, as amended, the U.K. Bribery Act 2010, or comparable foreign regulations; |
• | challenges enforcing or protecting our contractual and intellectual property rights, especially in those foreign countries that do not respect and protect intellectual property rights to the same extent as the United States or the United Kingdom; |
• | the impacts Brexit may have with respect to the cross-border acknowledgment of clinical trial results and marketing authorizations as well as recruitment of scientific personnel; |
• | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and |
• | business interruptions resulting from geo-political actions, including war and terrorism. |
• | the inability to recruit, train and retain adequate numbers of effective sales and marketing personnel; |
• | the inability of sales personnel to obtain access to physicians or persuade adequate numbers of physicians to prescribe any future product that we may develop; |
• | the lack of complementary treatments to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines; and |
• | unforeseen costs and expenses associated with creating an independent sales and marketing organization. |
• | the clinical indications for which our product candidates are approved; |
• | physicians, hospitals, cancer treatment centers, and patients considering our product candidates as a safe and effective treatment; |
• | hospitals and cancer treatment centers establishing the infrastructure required for the administration of the product candidate; |
• | the potential and perceived advantages of our product candidates over alternative treatments; |
• | the prevalence and severity of any side effects; |
• | product labeling or product insert requirements of the FDA, the EMA or other regulatory authorities; |
• | limitations or warnings contained in the labeling approved by the FDA or the EMA; |
• | the timing of market introduction of our product candidates as well as competitive products; |
• | the cost of treatment in relation to alternative treatments; |
• | the amount of upfront costs or training required for physicians to administer our product candidates; |
• | the pricing of our products and the availability of coverage and adequate reimbursement by third-party payors and government authorities; |
• | the willingness of patients to pay out-of-pocket in the absence of comprehensive coverage and adequate reimbursement by third-party payors and government authorities; |
• | relative convenience and ease of administration, including as compared to alternative treatments and competitive therapies; and |
• | the effectiveness of our sales and marketing efforts and distribution support. |
• | the impairment of our business reputation; |
• | the withdrawal of clinical trial participants; |
• | costs due to related litigation; |
• | the distraction of management’s attention from our primary business; |
• | substantial monetary awards to patients or other claimants; |
• | the inability to commercialize our product candidates; and |
• | decreased demand for our product candidates, if approved for commercial sale. |
• | collaborators have significant discretion in determining the efforts and resources that they will apply to these collaborations; |
• | collaborators may not perform their obligations as expected; |
• | collaborators may not pursue development and commercialization of any product candidates that achieve regulatory approval or may elect not to continue or renew development or commercialization programs based on clinical trial results, changes in the collaborators’ strategic focus or available funding, or external factors, such as an acquisition, that divert resources or create competing priorities; |
• | collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product candidate for clinical testing; |
• | collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our products or product candidates if the collaborators believe that competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours; this may also happen if the collaborators’ development of competing products is substantially faster than our development timelines; |
• | collaborators may not further develop product candidates developed by us or co-developed with us under the collaboration; |
• | product candidates discovered in collaboration with us may be viewed by our collaborators as competitive with their own product candidates or products, which may cause collaborators to cease to devote resources to the commercialization of our product candidates; |
• | a collaborator with marketing and distribution rights to one or more of our product candidates that achieve regulatory approval may not commit sufficient resources to the marketing and distribution of such product or products; |
• | disagreements with collaborators, including disagreements over proprietary rights, contract interpretation or the preferred course of development, might cause delays or termination of the research, development or commercialization of product candidates, might lead to additional responsibilities for us with respect to product candidates, or might result in litigation or arbitration, any of which would be time-consuming and expensive; |
• | collaborators have certain defined rights to change or expand the scope of development programs during the course of the collaboration. This may lead to additional research work for us that may be time-consuming and expensive. Such work may compete with our own development programs and may delay timelines to market or proof-of-concept for our product candidates. If development programs under the collaboration turn out to be more costly and time-consuming, such unanticipated costs and work could likewise compete with our internal development programs; |
• | collaborators may not properly maintain, enforce or defend our intellectual property or proprietary information or may use them in such a way as to invite litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential litigation; |
• | collaborators may infringe, misappropriate or otherwise violate the intellectual property or proprietary rights of third parties, which may expose us to litigation and potential liability, and collaborators may also allege that we are liable for potential infringement, misappropriation or other violations of third-party intellectual property or proprietary rights during the research and development work for the collaboration; |
• | certain collaborations may be terminated for the convenience of the collaborator and, if terminated, we could be required to raise additional capital to pursue further development or commercialization of the applicable product candidates. For example, certain of our collaboration and license agreements may be terminated for convenience upon the completion of a specified notice period; and |
• | collaborators may discontinue the development of product candidates within the collaboration, for example if they consider the results achieved so far or the product candidates not promising enough or if their development strategies change. |
• | have staffing difficulties; |
• | fail to comply with contractual obligations; |
• | experience regulatory compliance issues; |
• | undergo changes in priorities or become financially distressed; or |
• | form relationships with other entities, some of which may be our competitors. |
• | reliance on the third party for regulatory compliance and quality assurance; |
• | the possible breach of the manufacturing agreement by the third party; |
• | the possible misappropriation or unauthorized disclosure of our proprietary information, including our trade secrets and know-how; and |
• | the possible termination or nonrenewal of the agreement by the third party at a time that is costly or inconvenient for us. |
• | the scope of rights granted under the agreement and other interpretation-related issues; |
• | the extent to which our technology and processes infringe on intellectual property of the counterparty that is not subject to the agreement; |
• | the sublicensing of patent and other intellectual or proprietary rights under our collaborative development relationships; |
• | our diligence obligations under the agreement and what activities satisfy those diligence obligations; |
• | the inventorship and ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our counterparty and us and our partners; and |
• | the priority of invention of patented technology. |
• | others may be able to make products that are similar to our product candidates or utilize similar technology but that are not covered by the claims of the patents that we own or license now or in the future; |
• | we or our licensors or collaborators might not have been the first to make the inventions covered by the issued patents or pending patent applications that we own or license now or in the future; |
• | we or our licensors or collaborators might not have been the first to file patent applications covering certain of our or their inventions; |
• | others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights or any intellectual property rights we may license; |
• | it is possible that our present or future pending patent applications (whether owned or licensed) will not lead to issued patents; |
• | it is possible that there are or will be prior public disclosures that could invalidate our or our licensors’ or collaboration partners’ patents; |
• | issued patents that we hold rights to may fail to provide us with any competitive advantage, or may be held invalid or unenforceable, including as a result of legal challenges by our competitors or other third parties; |
• | our competitors or other third parties might conduct research and development activities in countries where we do not have patent rights or in countries where research and development safe harbor laws exist, and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; |
• | we may not develop additional proprietary technologies that are patentable; |
• | the ownership, validity or enforceability of our patents or patent applications may be challenged by third parties; |
• | the patents or pending or future applications of others, if issued, may harm our business; and |
• | we may choose not to file a patent in order to maintain certain trade secrets or know-how, and a third party may subsequently file a patent covering such intellectual property. |
• | the FDA or comparable foreign regulatory authorities may disagree with the design or implementation of our or our collaborators’ clinical trials; |
• | we or our collaborators may be unable to demonstrate to the satisfaction of the FDA or comparable foreign regulatory authorities that our product candidates are safe, pure, potent and have a favorable risk/benefit profile for any of their proposed indications; |
• | the results of clinical trials may not meet the level of statistical significance required by the FDA or comparable foreign regulatory authorities for approval; |
• | the FDA or comparable foreign regulatory authorities may disagree with our interpretation of data from pre-clinical programs or clinical trials; |
• | data collected from clinical trials of product candidates may not be sufficient to the satisfaction of the FDA or comparable foreign regulatory authorities to support the submission of a BLA or other comparable submission in foreign jurisdictions or to obtain regulatory approval in the United States or elsewhere; |
• | the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval. |
• | restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, or voluntary or mandatory product recalls; |
• | clinical trial holds; |
• | fines, warning letters or other regulatory enforcement action; |
• | refusal by the FDA to approve pending applications or supplements to approved applications filed by us; |
• | product seizure or detention, or refusal to permit the import or export of products; and |
• | injunctions or the imposition of civil or criminal penalties. |
• | the demand for our current or future product candidates, if we obtain regulatory approval; |
• | our ability to set a price that we believe is fair for our products; |
• | our ability to obtain coverage and adequate reimbursement for a product; |
• | our ability to generate revenue and achieve or maintain profitability; |
• | the level of taxes that we are required to pay; and |
• | the availability of capital. |
• | the federal Anti-Kickback Statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under federal and state healthcare programs such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation; |
• | the federal civil and criminal false claims and civil monetary penalties laws, including the federal False Claims Act, or FCA, imposes criminal and civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government. In addition, the government may assert that a claim including items and services resulting from a violation of the federal Anti-Kickback Statute constitutes a false of fraudulent claim for purposes of the False Claims Act; |
• | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program, or knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services; similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation; |
• | the federal physician payment transparency requirements, sometimes referred to as the “Sunshine Act” under the Affordable Care Act, require manufacturers of drugs, devices, biologics and medical supplies that are reimbursable under Medicare, Medicaid, or the Children’s Health Insurance Program to report to the Department of Health and Human Services information related to transfers of value made to physicians (currently defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, as well as ownership and investment interests of such physicians and their immediate family members. Effective January 1, 2022, these reporting obligations will extend to include transfers of value made to certain non-physician providers including physician assistants, nurse practitioners, clinical nurse specialists, anesthesiologist assistants, certified registered nurse anesthetists and certified nurse midwives during the previous year; |
• | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 and its implementing regulations, impose obligations on certain covered entity healthcare providers, health plans, and healthcare clearinghouses as well as their business associates that perform certain services involving the use or disclosure of individually identifiable health information and their |
• | analogous state laws and regulations, such as state anti-kickback and false claims laws may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers. Some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government in addition to requiring drug manufacturers to report information related to payments to physicians and other healthcare providers or marketing expenditures. Further, many state laws governing the privacy and security of health information in certain circumstances, differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
• | convey, sell, lease, transfer, assign, dispose of or otherwise make cash payments consisting of all or any part of our business or property; |
• | effect certain changes in our business, management, ownership or business locations; |
• | merge or consolidate with, or acquire all or substantially all of the capital stock or assets of, any other company; |
• | create, incur, assume or be liable for any additional indebtedness, or create, incur, allow or permit to exist any additional liens; |
• | pay cash dividends on, make any other distributions in respect of, or redeem, retire or repurchase, any shares of our capital stock; |
• | make certain investments; and |
• | enter into transactions with our affiliates. |
• | increased operating expenses and cash requirements; |
• | the assumption of additional indebtedness or contingent liabilities; |
• | assimilation of operations, intellectual property and products of an acquired company or product, including difficulties associated with integrating new personnel; |
• | the diversion of our management’s attention from our existing product programs and initiatives in pursuing such a strategic merger or acquisition; |
• | retention of key employees, the loss of key personnel, and uncertainties in our ability to maintain key business relationships; |
• | risks and uncertainties associated with the other party to such a transaction, including the prospects of that party and their existing products or product candidates and regulatory approvals; and |
• | our inability to generate revenue from acquired technology and/or products sufficient to meet our objectives in undertaking the acquisition or even to offset the associated acquisition and maintenance costs. |
• | economic weakness, including inflation, or political instability in particular non-U.S. economies and markets; |
• | differing and changing regulatory requirements for product approvals; |
• | differing jurisdictions could present different issues for securing, maintaining or obtaining freedom to operate in such jurisdictions; |
• | potentially reduced protection for intellectual property and proprietary rights; |
• | difficulties in compliance with different, complex and changing laws, regulations and court systems of multiple jurisdictions and compliance with a wide variety of foreign laws, treaties and regulations; |
• | changes in non-U.S. regulations and customs, tariffs and trade barriers; |
• | changes in non-U.S. currency exchange rates of the pound sterling, U.S. dollar, euro and currency controls; |
• | changes in a specific country’s or region’s political or economic environment, including the implications of the recent decision of the United Kingdom to withdraw from the European Union; |
• | trade protection measures, import or export licensing requirements or other restrictive actions by governments; |
• | differing reimbursement regimes and price controls in certain non-U.S. markets; |
• | negative consequences from changes in tax laws; |
• | compliance with tax, employment, immigration and labor laws for employees living or traveling abroad, including, for example, the variable tax treatment in different jurisdictions of options granted under our share option schemes or equity incentive plans; |
• | workforce uncertainty in countries where labor unrest is more common than in the United States; |
• | litigation or administrative actions resulting from claims against us by current or former employees or consultants individually or as part of class actions, including claims of wrongful terminations, discrimination, misclassification or other violations of labor law or other alleged conduct; |
• | difficulties associated with staffing and managing international operations, including differing labor relations; |
• | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and |
• | business interruptions resulting from geo-political actions, including war and terrorism, or natural disasters including earthquakes, typhoons, floods and fires. |
• | adverse results or delays in pre-clinical studies or clinical trials; |
• | reports of adverse events in products similar or perceived to be similar to those we are developing or clinical trials of such products; |
• | an inability to obtain additional funding; |
• | failure by us to successfully develop and commercialize our product candidates; |
• | failure by us to maintain our existing strategic collaborations or enter into new collaborations; |
• | failure by us to identify additional product candidates for our pipeline; |
• | failure by us or our licensors and strategic partners to prosecute, maintain, protect or enforce our intellectual property and proprietary rights; |
• | disputes or other developments relating to intellectual and other proprietary rights, including litigation |
• | matters and our ability to obtain patent and other intellectual property protection for our technologies; |
• | changes in laws or regulations applicable to future products; |
• | an inability to obtain adequate product supply for our product candidates or the inability to do so at acceptable prices; |
• | adverse regulatory decisions; |
• | the introduction of new products, services or technologies by our competitors; |
• | failure by us to meet or exceed financial projections we may provide to the public; |
• | failure by us to meet or exceed the financial projections of the investment community; |
• | the perception of the pharmaceutical industry by the public, legislatures, regulators and the investment community; |
• | changes in the structure of healthcare payment systems; |
• | announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments by us, our strategic partner or our competitors; |
• | additions or departures of key scientific or management personnel; |
• | significant lawsuits, including patent or shareholder litigation; |
• | changes in the market valuations of similar companies; |
• | general economic, industry, political and market conditions, including, but not limited to, the ongoing impact of the COVID-19 pandemic; |
• | sales of our ADSs or ordinary shares by us or our shareholders in the future; and |
• | the trading volume of our ADSs. |
• | delaying, deferring, or preventing a change in control; |
• | entrenching our management and/or the board of directors; |
• | impeding a merger, scheme of arrangement, takeover, or other business combination involving us; or |
• | discouraging a potential acquirer from making a takeover offer or otherwise attempting to obtain control of us. |
• | not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, or Section 404; |
• | not being required to comply with any requirement that has or may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements; |
• | being permitted to provide only two years of audited financial statements in this initial registration statement, in addition to any required unaudited interim financial statements, with correspondingly reduced “Management’s Discussion and Analysis of Financial Condition and Results of Operations” disclosure; |
• | reduced disclosure obligations regarding executive compensation; and |
• | an exemption from the requirement to seek nonbinding advisory votes on executive compensation or golden parachute arrangements. |
• | In connection with a potential offer, if following an approach by or on behalf of a potential bidder, the company is “the subject of rumor or speculation” or there is an “untoward movement” in the company’s share price, there is a requirement for the potential bidder to make a public announcement about a potential offer for the company, or for the company to make a public announcement about its review of a potential offer. |
• | When a person or group of persons acting in concert (a) acquires, whether by a series of transactions over a period of time or not, interests in shares carrying 30% or more of the voting rights of a company (which percentage is treated by the Takeover Code as the level at which effective control is obtained) or (b) acquires an interest in any other shares which increases the percentage of shares carrying voting rights in which they are interested when they are already interested in shares which carry not less than 30% of the voting rights but do not hold shares carrying more than 50% of such voting rights, they must make a cash offer to all other shareholders at the highest price paid by them or any person acting in concert with them in the 12 months before the offer was announced. |
• | When interests in shares carrying 10% or more of the voting rights of a class have been acquired by an offeror (i.e., a bidder) and any person acting in concert with it in the offer period (i.e., before the shares subject to the offer have been acquired) or within the previous 12 months, the offer must be in cash or be accompanied by a cash alternative for all shareholders of that class at the highest price paid by the offeror or any person acting in concert with them in that period. Further, if an offeror or any person acting in concert with them acquires any interest in shares during the offer period, the offer for the shares must be in cash or accompanied by a cash alternative at a price at least equal to the price paid for such shares during the offer period. |
• | If after an announcement is made, the offeror or any person acting in concert with them acquires an interest in shares in an offeree company (i.e., a target) at a price higher than the value of the offer, the offer must be increased to not less than the highest price paid for the interest in shares so acquired. |
• | The board of directors of the offeree company must appoint a competent independent adviser whose advice on the financial terms of the offer must be made known to all the shareholders, together with the opinion of the board of directors of the offeree company. |
• | Special or favorable deals for selected shareholders are not permitted, except in certain circumstances where independent shareholder approval is given and the arrangements are regarded as fair and reasonable in the opinion of the financial adviser to the offeree. |
• | All shareholders must be given the same information. |
• | Each document published in connection with an offer by or on behalf of the offeror or offeree must state that the directors of the offeror or the offeree, as the case may be, accept responsibility for the information contained therein. |
• | Profit forecasts, quantified financial benefits statements and asset valuations must be made to specified standards and must be reported on by professional advisers. |
• | Misleading, inaccurate or unsubstantiated statements made in documents or to the media must be publicly corrected immediately. |
• | Actions during the course of an offer by the offeree company, which might frustrate the offer are generally prohibited unless shareholders approve these plans. Frustrating actions would include, for example, lengthening the notice period for directors under their service contract or agreeing to sell off material parts of the target group. |
• | Stringent and detailed requirements are laid down for the disclosure of dealings in relevant securities during an offer, including the prompt disclosure of positions and dealing in relevant securities by the parties to an offer and any person who is interested (directly or indirectly) in 1% or more of any class of relevant securities. |
• | Employees of both the offeror and the offeree company and the trustees of the offeree company’s pension scheme must be informed about an offer. In addition, the offeree company’s employee representatives and pension scheme trustees have the right to have a separate opinion on the effects of the offer on employment appended to the offeree board of directors’ circular or published on a website. |
• | under our articles of association to be effective upon completion of this offering, any resolution put to the vote of a general meeting must be decided exclusively on a poll. Under English law, it would be possible for our articles of association to be amended such that each shareholder present at a meeting has only one vote unless demand is made for a vote on a poll, in which case each holder gets one vote per share owned. Under U.S. law, each shareholder typically is entitled to one vote per share at all meetings; |
• | under English law, subject to certain exceptions and disapplications, each shareholder generally has preemptive rights to subscribe on a proportionate basis to any issuance of ordinary shares or rights to subscribe for, or to convert securities into, ordinary shares for cash. Under U.S. law, shareholders generally do not have preemptive rights unless specifically granted in the certificate of incorporation or otherwise; |
• | under English law and our articles of association, certain matters require the approval of 75% of the shareholders who vote (in person or by proxy) on the relevant resolution (or on a poll of shareholders representing 75% of the ordinary shares voting (in person or by proxy)), including amendments to the articles of association. This may make it more difficult for us to complete corporate transactions deemed advisable by our board of directors. Under U.S. law, generally only majority shareholder approval is required to amend the certificate of incorporation or to approve other significant transactions; |
• | in the United Kingdom, takeovers may be structured as takeover offers or as schemes of arrangement. Under English law, a bidder seeking to acquire us by means of a takeover offer would need to make an offer for all of our outstanding ordinary shares/ADSs. If acceptances are not received for 90% or more of the ordinary shares/ADSs under the offer, under English law, the bidder cannot complete a “squeeze out” to obtain 100% control of us. Accordingly, acceptances of 90% of our outstanding ordinary shares/ADSs will likely be a condition in any takeover offer to acquire us, not 50% as is more common in tender offers for corporations organized under Delaware law. By contrast, a scheme of arrangement, the successful completion of which would result in a bidder obtaining 100% control of us, requires the approval of a majority of shareholders voting at the meeting and representing 75% of the ordinary shares voting for approval; |
• | under English law and our articles of association, shareholders and other persons whom we know or have reasonable cause to believe are, or have been, interested in our shares may be required to disclose information regarding their interests in our shares upon our request, and the failure to provide the required information could result in the loss or restriction of rights attaching to the shares, including prohibitions on certain transfers of the shares, withholding of dividends and loss of voting rights. Comparable provisions generally do not exist under U.S. law; and |
• | the quorum requirement for a shareholders’ meeting is a minimum of two shareholders entitled to vote at the meeting and present in person or by proxy or, in the case of a shareholder which is a corporation, represented by a duly authorized representative. Under U.S. law, a majority of the shares eligible to vote must generally be present (in person or by proxy) at a shareholders’ meeting in order to constitute a quorum. The minimum number of shares required for a quorum can be reduced pursuant to a provision in a company’s certificate of incorporation or bylaws, but typically not below one-third of the shares entitled to vote at the meeting. |
• | the initiation, timing, progress and results of our current and future preclinical studies and clinical trials and related preparatory work and the period during which the results of the trials will become available, as well as our research and development programs; |
• | our estimates regarding expenses, future revenue, capital requirements and needs for additional financing; |
• | our expectations regarding timing of regulatory filings for, or our ability to obtain regulatory approval of, tebentafusp or any of our other product candidates; |
• | our ability to identify and develop additional product candidates using our ImmTAX platform; |
• | business disruptions affecting the initiation, patient enrollment, development and operation of our clinical trials, including a public health emergency, such as the ongoing the coronavirus 2019, or COVID-19, pandemic; |
• | the potential benefits of our product candidates; |
• | our expectations regarding the potential commercialization of, the potential market size and the rate and degree of market acceptance for any product candidates that we develop; |
• | our business strategies and goals; |
• | our plans to collaborate, or statements regarding our current collaborations; |
• | our ability to find future partners and collaborators; |
• | the performance of our third-party suppliers and manufacturers, |
• | our expectations regarding our ability to obtain, maintain and enforce intellectual property protection for our product candidates and our ability to operate our business without infringing, misappropriating or otherwise violating the intellectual property rights of others; |
• | the effects of competition with respect to tebentafusp or any of our other current or future product candidates, as well as innovations by current and future competitors in our industry; |
• | our financial performance and our ability to effectively manage our anticipated growth; |
• | our ability to identify, recruit and retain key personnel; and |
• | our expectations regarding the uses of the proceeds from this offering and the sufficiency of such net proceeds together with our existing cash and cash equivalents to fund our operations and capital expenditures. |
• | approximately $100 million to fund tebentafusp, our lead ImmTAC, for the treatment of metastatic uveal melanoma through the completion of our Phase 3 clinical trial as well as preparations for a commercial launch; |
• | approximately $20.0 million to advance the clinical development of IMC-C103C targeting MAGE A4 for the treatment of solid tumors; |
• | approximately $20.0 million to advance the clinical development of IMC-F106C targeting PRAME for the treatment of solid tumors; |
• | approximately $10.0 million to advance the clinical development of IMC-I109V targeting a functional cure for chronic HBV; |
• | approximately $20.0 million to continue to advance our pre-clinical programs and invest in our ImmTAX platform to discover and develop novel therapeutics; and |
• | the remainder for working capital and general corporate purposes. |
• | an actual basis; |
• | a pro forma basis to give effect to (i) the issuance and sale of 823,719 Series C preferred shares for aggregate gross proceeds of $75.0 million in December 2020; (ii) the borrowing of $50.0 million in November 2020 under a new debt facility with Oxford Finance Luxembourg S.A.R.L., or Oxford Finance; and (iii) our corporate reorganization; and |
• | a pro forma as adjusted basis to give further effect to (i) the sale of 8,333,333 ADSs in this offering at an assumed initial public offering price of $24.00 per ADS, which is the midpoint of the price range set forth on the cover page of this prospectus, and after deducting the underwriting discounts and commissions and estimated offering expenses payable by us; and (ii) the issuance and sale by us in the concurrent private placement of $15.0 million of our ADSs, or an estimated 625,000 ADSs based on the assumed initial public offering price, to the Bill & Melinda Gates Foundation. |
| | | As of September 30, 2020 | ||||||||||
| | | Actual £ | | | Actual $(1) | | | Pro Forma | | | Pro Forma As Adjusted | |
| | | (in thousands except share and per share amounts) | ||||||||||
Cash and cash equivalents | | | £56,687 | | | $73,245 | | | $198,245 | | | $394,845 |
Interest-bearing loans and borrowings | | | | | | | 50,000 | | | 50,000 | ||
Shareholders’ equity: | | | | | | | | | ||||
Series A preferred shares, nominal value £0.0001 per share, 1,699,576 shares issued and outstanding, actual; no shares issued and outstanding, pro forma and pro forma as adjusted | | | — | | | — | | | — | | | — |
Series B preferred shares, nominal value £0.0001 per share, 1,148,703 shares issued and outstanding, actual; no shares issued and outstanding, pro forma and pro forma as adjusted | | | — | | | — | | | — | | | — |
Series C preferred shares, nominal value £0.0001 per share, 823,719 shares issued and outstanding, actual; no shares issued and outstanding, pro forma and pro forma as adjusted | | | — | | | — | | | — | | | — |
G shares, nominal value £0.0001 per share, 62,750 shares issued and outstanding, actual; no shares issued and outstanding, pro forma and pro forma as adjusted | | | — | | | — | | | — | | | — |
Voting ordinary shares, 2,551,624 shares issued and outstanding, actual; 2,551,624 shares issued and outstanding, pro forma; shares authorized, shares issued and outstanding, pro forma as adjusted | | | 1 | | | 1 | | | 3 | | | 4 |
Non-voting ordinary shares, nominal value £0.0001 per share, 831,628 shares issued and outstanding, actual; no shares issued and outstanding, pro forma and pro forma as adjusted | | | — | | | — | | | — | | | — |
Additional paid-in capital | | | 330,390 | | | 426,897 | | | 501,893 | | | 698,493 |
Other reserves | | | 16,146 | | | 20,863 | | | 20,863 | | | 20,863 |
Accumulated deficit | | | (330,989) | | | (427,671) | | | (427,671) | | | (427,671) |
Total shareholders’ equity | | | 15,548 | | | 20,090 | | | 95,089 | | | 291,689 |
Total capitalization | | | £15,548 | | | $20,090 | | | $145,089 | | | $341,689 |
(1) | Translated solely for convenience of the reader into U.S. dollars at the rate of £1.00 to $1.2921, which was the noon buying rate of the Federal Reserve Bank of New York on September 30, 2020. |
• | 832,904 ordinary shares issuable upon the exercise of options outstanding under our existing equity incentive plans as of September 30, 2020, with a weighted-average exercise price of £63.23 per share; and |
• | 5,706,279 ordinary shares reserved for future issuance under our 2021 EIP which will become effective in connection with this offering, as well as any automatic annual increases in the number of ordinary shares reserved for future issuance under the 2021 EIP, as more fully described in the section titled “Management—Equity Incentive Plans;” and |
• | 4,014,267 ordinary shares (assuming an initial public offering price of $24.00 per ADS, which is the midpoint of the price range set forth on the cover page of this prospectus) underlying the grants to be issued prior to the closing of this offering to certain of our officers, directors and employees under our 2021 EIP, contingent and effective upon the execution and delivery of the underwriting agreement relating to this offering, with an exercise price that is equal to or greater than the price per ADS at which our ADSs are first sold to the public in this offering. |
Assumed initial public offering price per ADS | | | | | $24.00 | |
Historical net tangible book value per ADS as of September 30, 2020 | | | $3.68 | | | |
Decrease in net tangible book value per ADS attributable to our corporate reorganization and Series C preferred share issuance | | | (0.69) | | | |
Pro forma net tangible book value per ADS as of September 30, 2020 | | | 2.99 | | | |
Increase in net tangible book value per ADS attributable to this offering and concurrent private placement | | | 4.17 | | | |
Pro forma as adjusted net tangible book value per ADS after this offering and concurrent private placement | | | | | 7.16 | |
Dilution in as adjusted net tangible book value per ADS to new investors participating in this offering | | | | | $16.84 |
| | | Ordinary Shares or ADSs Purchased | | | Total Consideration | | | Average Price Per Ordinary Share | | | Average Price Per ADS | |||||||
| | | Number | | | Percent | | | Amount | | | Percent | | | | | |||
Existing shareholders | | | 31,782,885 | | | 79.2% | | | $330,391,000 | | | 62.3% | | | $10.40 | | | $10.40 |
New investors | | | 8,333,333 | | | 20.8% | | | $200,000,000 | | | 37.7% | | | $24.00 | | | $24.00 |
Totals | | | 40,116,218 | | | 100.0% | | | $530,391,000 | | | 100.0% | | | $13.22 | | | $13.22 |
• | 832,904 ordinary shares issuable upon the exercise of options outstanding under our existing equity incentive plans as of September 30, 2020, with a weighted-average exercise price of £63.23 per share; and |
• | 5,706,279 ordinary shares reserved for future issuance under our 2021 EIP which will become effective in connection with this offering, as well as any automatic annual increases in the number of ordinary shares reserved for future issuance under the 2021 EIP, as more fully described in the section titled “Management—Equity Incentive Plans;” and |
• | 4,014,267 ordinary shares (assuming an initial public offering price of $24.00 per ADS, which is the midpoint of the price range set forth on the cover page of this prospectus) underlying the grants to be issued prior to the closing of this offering to certain of our officers, directors and employees under our 2021 EIP, contingent and effective upon the execution and delivery of the underwriting agreement relating to this offering, with an exercise price that is equal to or greater than the price per ADS at which our ADSs are first sold to the public in this offering. |
| | | For the nine-month period ended September 30, | | | For the years ended December 31, | |||||||
| | | 2020 | | | 2019 | | | 2019 | | | 2018 | |
| | | (pounds sterling, in thousands, except for share and per share data) | | | (pounds sterling, in thousands, except for share and per share data) | |||||||
Consolidated statement of loss and other comprehensive income data: | | | | | | | | | ||||
Revenue | | | 22,694 | | | 20,027 | | | 25,669 | | | 23,654 |
Other operating income | | | 408 | | | 420 | | | 185 | | | 622 |
Operating expenses: | | | | | | | | | ||||
Research and development | | | (57,566) | | | (75,415) | | | (99,991) | | | (83,575) |
Administrative expenses | | | (31,569) | | | (35,611) | | | (44,183) | | | (34,156) |
Operating loss | | | (66,033) | | | (90,579) | | | (118,320) | | | (93,455) |
Other income | | | — | | | — | | | — | | | 4,979 |
Finance income | | | 1,972 | | | 1,134 | | | 1,510 | | | 1,140 |
Finance costs | | | (2,272) | | | (6,532) | | | (9,379) | | | (842) |
Non-operating (expense) / income | | | (300) | | | (5,398) | | | (7,869) | | | 5,277 |
Loss before tax | | | (66,333) | | | (95,977) | | | (126,189) | | | (88,178) |
Income tax credit | | | 11,120 | | | 18,011 | | | 22,258 | | | 16,548 |
Loss for the period | | | (55,213) | | | (77,966) | | | (103,931) | | | (71,630) |
Exchange differences on translation of foreign operations | | | 338 | | | 82 | | | (99) | | | 72 |
Income tax effect relating to the components of other comprehensive income | | | — | | | — | | | — | | | 3,634 |
Total comprehensive loss for the period, net of tax | | | (54,875) | | | (77,884) | | | (104,030) | | | (67,924) |
| | | | | | | | | |||||
Pro forma per share data:(1) | | | | | | | | | ||||
Basic and diluted loss per share(2) | | | (0.002) | | | (0.004) | | | (0.005) | | | (0.003) |
Basic and diluted weighted average number of shares | | | 27,306,935 | | | 21,579,880 | | | 22,297,935 | | | 21,558,890 |
| | | As of September 30, | | | As of December 31, | ||||
| | | 2020 | | | 2019 | | | 2018 | |
| | | (pounds sterling, in thousands) | | | (pounds sterling, in thousands) | ||||
Consolidated statement of financial position data: | | | | | | | |||
Cash and cash equivalents(3) | | | 56,687 | | | 73,966 | | | 124,385 |
Working capital(4) | | | 29,335 | | | 39,768 | | | 121,574 |
Total assets | | | 130,839 | | | 185,649 | | | 195,777 |
Interest-bearing loans and borrowings(3) | | | — | | | 19,157 | | | 18,878 |
Total liabilities(2) | | | 115,291 | | | 170,878 | | | 139,195 |
Share capital | | | 1 | | | — | | | — |
Total equity | | | 15,548 | | | 14,771 | | | 56,582 |
(1) | The unaudited pro forma loss per share data gives effect to our corporate reorganization. |
(2) | See Note 10 to our audited consolidated financial statements for the year ended December 31, 2019 and year ended December 31, 2018 and Note 6 to our unaudited consolidated financial statements appearing elsewhere in this prospectus for a description of the method used to compute diluted net loss per share. |
(3) | Excludes the borrowing of $50.0 million in November 2020 under a new debt facility with Oxford Finance Luxembourg S.A.R.L. |
(4) | We define working capital as current assets less current liabilities. |
• | after reviewing trial results, our collaboration partners may abandon projects that might previously have been believed to be promising; |
• | we, our collaboration partners, or regulators may suspend or terminate clinical trials if the participating subjects or patients are being exposed to unacceptable health risks; |
• | our potential products may not have the desired effects or may include undesirable side effects or other characteristics that preclude regulatory approval or limit their commercial use if approved; |
• | manufacturers may not meet the necessary standards for the production of the product candidates or may not be able to supply the product candidates in a sufficient quantity; |
• | regulatory authorities may find that our clinical trial design or conduct does not meet the applicable approval requirements; and |
• | safety and efficacy results in various human clinical trials reported in scientific and medical literature may not be indicative of results we obtain in our clinical trials. |
| | | Nine months ended September 30, | |||||||
| | | 2020 | | | 2019 | ||||
| | | $000 | | | £000 | | | £000 | |
| | | (unaudited) | |||||||
Revenue | | | 29,323 | | | 22,694 | | | 20,027 |
Research and development expenses | | | (74,381) | | | (57,566) | | | (75,415) |
Administrative expenses | | | (40,790) | | | (31,569) | | | (35,611) |
Other operating income | | | 527 | | | 408 | | | 420 |
Operating loss | | | (85,321) | | | (66,033) | | | (90,579) |
Other income | | | — | | | — | | | — |
Finance income | | | 2,548 | | | 1,972 | | | 1,134 |
Finance costs | | | (2,936) | | | (2,272) | | | (6,532) |
Non-operating (expense) / income | | | (388) | | | (300) | | | (5,398) |
Loss before taxes | | | (85,709) | | | (66,333) | | | (95,977) |
Income tax credit | | | 14,368 | | | 11,120 | | | 18,011 |
Loss for the period | | | (71,341) | | | (55,213) | | | (77,966) |
| | | Nine months ended September 30, | |||||||
| | | 2020 | | | 2019 | ||||
| | | $000 | | | £000 | | | £000 | |
| | | (unaudited) | |||||||
GSK | | | 5,613 | | | 4,344 | | | 3,796 |
Eli Lilly | | | 4,551 | | | 3,522 | | | 1,886 |
Genentech | | | 19,159 | | | 14,828 | | | 14,345 |
Total | | | 29,323 | | | 22,694 | | | 20,027 |
| | | Nine months ended September 30, | |||||||
| | | 2020 | | | 2019 | ||||
| | | $000 | | | £000 | | | £000 | |
| | | (unaudited) | |||||||
External research and development expenses: | | | | | | | |||
Tebentafusp | | | 34,759 | | | 26,901 | | | 42,035 |
IMC-F106C (PRAME) | | | 1,620 | | | 1,254 | | | 2,264 |
IMC-C103C (MAGE-A4) | | | 4,654 | | | 3,602 | | | 2,284 |
Other programs | | | 8,471 | | | 6,556 | | | 4,653 |
Research expenses | | | 536 | | | 415 | | | 567 |
Total external research and development expenses | | | 50,040 | | | 38,728 | | | 51,803 |
Internal research and development expenses: | | | | | | | |||
Headcount related expenses | | | 18,510 | | | 14,325 | | | 16,895 |
Laboratory consumables | | | 4,199 | | | 3,250 | | | 5,123 |
Laboratory equipment expenses | | | 1,552 | | | 1,201 | | | 1,231 |
Other | | | 80 | | | 62 | | | 363 |
Total internal research and development expenses | | | 24,341 | | | 18,838 | | | 23,612 |
Total research and development expenses | | | 74,381 | | | 57,566 | | | 75,415 |
| | | Year ended December 31, | ||||
| | | 2019 | | | 2018 | |
| | | £000 | | | £000 | |
Revenue | | | 25,669 | | | 23,654 |
Research and development expenses | | | (99,991) | | | (83,575) |
Administrative expenses | | | (44,183) | | | (34,156) |
Other operating income | | | 185 | | | 622 |
Operating loss | | | (118,320) | | | (93,455) |
Other income | | | — | | | 4,979 |
Finance income | | | 1,510 | | | 1,140 |
Finance costs | | | (9,379) | | | (842) |
Non-operating (expense) / income | | | (7,869) | | | 5,277 |
Loss before taxes | | | (126,189) | | | (88,178) |
Income tax credit | | | 22,258 | | | 16,548 |
Net loss | | | (103,931) | | | (71,630) |
| | | Year ended December 31, | ||||
| | | 2019 | | | 2018 | |
| | | £000 | | | £000 | |
GSK | | | 5,753 | | | 6,079 |
Eli Lilly | | | 819 | | | 8,561 |
Genentech | | | 19,097 | | | 1,461 |
MedImmune | | | — | | | 7,553 |
Total | | | 25,669 | | | 23,654 |
| | | Year ended December 31, | ||||
| | | 2019 | | | 2018 | |
| | | £000 | | | £000 | |
External research and development expenses: | | | | | ||
Tebentafusp | | | 52,406 | | | 34,493 |
IMC-F106C (PRAME) | | | 2,825 | | | 1,179 |
IMC-C103C (MAGE-A4) | | | 3,182 | | | 2,315 |
Other programs | | | 8,870 | | | 9,710 |
Research expenses | | | 795 | | | 1,025 |
Total external research and development expenses | | | 68,078 | | | 48,722 |
| | | Year ended December 31, | ||||
| | | 2019 | | | 2018 | |
| | | £000 | | | £000 | |
Internal research and development expenses: | | | | | ||
Headcount related expenses | | | 23,320 | | | 23,475 |
Laboratory consumables | | | 6,704 | | | 8,146 |
Laboratory equipment expenses | | | 1,411 | | | 1,589 |
Other | | | 478 | | | 1,643 |
Total internal research and development expenses | | | 31,913 | | | 34,853 |
Total research and development expenses | | | 99,991 | | | 83,575 |
| | | Nine months ended September 30, | | | Year ended December 31, | ||||||||||
| | | 2020 | | | 2020 | | | 2019 | | | 2019 | | | 2018 | |
| | | $000 | | | £000 | | | £000 | | | £000 | | | £000 | |
| | | (unaudited) | | | | | | | | | |||||
Brought forward | | | 95,572 | | | 73,966 | | | 124,385 | | | 124,385 | | | 82,883 |
Net cash used in operating activities | | | (51,683) | | | (39,998) | | | (75,690) | | | (101,376) | | | (16,626) |
Net cash (used in) / provided by investing activities | | | (1,739) | | | (1,346) | | | (2,543) | | | (4,137) | | | 58,014 |
Net cash provided by financing activities | | | 30,983 | | | 23,978 | | | 56,172 | | | 55,127 | | | 101 |
Foreign exchange on cash held | | | 112 | | | 87 | | | 74 | | | (33) | | | 13 |
Cash and cash equivalents | | | 73,245 | | | 56,687 | | | 102,398 | | | 73,966 | | | 124,385 |
• | continue to advance the development of our clinical trials and pre-clinical programs; |
• | continue to invest in our soluble TCR platforms to conduct research to identify novel technologies; |
• | change or add additional suppliers; |
• | add additional infrastructure to our quality control, quality assurance, legal, compliance and other groups to support our operations as we progress product candidates toward commercialization; |
• | seek to attract and retain skilled personnel; |
• | create additional infrastructure to support our operations as a public company listed in the United States and our product development and planned future commercialization efforts; |
• | seek marketing approvals and reimbursement for our product candidates; |
• | establish a sales, marketing and distribution infrastructure to commercialize any products for which we may obtain marketing approval; |
• | seek to identify and validate additional product candidates; |
• | acquire or in-license other product candidates and technologies; |
• | maintain, protect, defend, enforce and expand our intellectual property portfolio; and |
• | experience any delays, interruptions or encounter issues with any of the above, including any delays or other impacts as a result of the COVID-19 pandemic. |
• | progress, timing, scope and costs of our clinical trials, including the ability to timely initiate clinical sites, enroll subjects and manufacture soluble bispecific TCR product candidates for our ongoing, planned and potential future clinical trials; |
• | time and costs required to perform research and development to identify and characterize new product candidates from our research programs; |
• | time and cost necessary to obtain regulatory authorizations and approvals that may be required by regulatory authorities to execute clinical trials or commercialize our products; |
• | our ability to successfully commercialize our product candidates, if approved; |
• | our ability to have clinical and commercial products successfully manufactured consistent with FDA, EMA and other authorities’ regulations; |
• | amount of sales and other revenues from product candidates that we may commercialize, if any, including the selling prices for such potential products and the availability of adequate third-party coverage and reimbursement for patients; |
• | sales and marketing costs associated with commercializing our products, if approved, including the cost and timing of building our marketing and sales capabilities; |
• | cost of building, staffing and validating our manufacturing processes, which may include capital expenditure; |
• | terms and timing of any revenue from our existing collaborations; |
• | costs of operating as a public company; |
• | time and cost necessary to respond to technological, regulatory, political and market developments; |
• | costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; |
• | costs, associated with, and terms and timing of, any future any potential acquisitions, strategic collaborations, licensing agreements or other arrangements that we may establish; and |
• | inability of clinical sites to enroll patients as healthcare capacities are required to cope with natural disasters, epidemics or other health system emergencies, such as the COVID-19 pandemic. |
As at September 30, 2020 | | | Less than 1 year | | | 1-3 years | | | 3-5 years | | | More than 5 years | | | Total |
Lease liabilities – existing | | | 4,275 | | | 7,910 | | | 6,548 | | | 35,657 | | | 54,390 |
Lease liabilities – contingent | | | — | | | 2,254 | | | 2,471 | | | 1,841 | | | 6,566 |
Manufacturing | | | 2,021 | | | 471 | | | — | | | — | | | 2,492 |
Capital commitments | | | 2,134 | | | — | | | — | | | — | | | 2,134 |
Total contractual obligations (in thousands, pounds) | | | 8,430 | | | 10,635 | | | 9,019 | | | 37,498 | | | 65,582 |
Total contractual obligations (in thousands, U.S. dollars) | | | 10,373 | | | 13,741 | | | 11,653 | | | 48,451 | | | 84,737 |
• | we have key worker status which allows continuity of providing services throughout a prolonged lockdown period; |
• | we have a track record of meeting expectations under its collaboration agreements and meeting expected milestones within the contracted timeframe; |
• | we have a history of being able to access equity and loan financing as and when needed; and |
• | we have the ability and history to control capital expenditure costs and lower other operational spend, as necessary. |
• | whether achievement of a development milestone is highly susceptible to factors outside the entity’s influence, such as milestones involving the judgment or actions of third parties, including regulatory bodies or the customer; |
• | whether the uncertainty about the achievement of the milestone is not expected to be resolved for a long period of time; |
• | whether we can reasonably predict that a milestone will be achieved based on previous experience; and. |
• | the complexity and inherent uncertainty underlying the achievement of the milestone. |
• | adjustments arising from a change in the estimate of when the performance obligation will have been completed. |
• | adjustment to revenue that affects deferred revenue; |
• | a change in the estimate of the transaction price due to changes in the assessment of whether variable consideration is constrained because it is not considered probable of being received; and |
• | the recognition of revenue. |
• | past history and experience with similar contracts. |
• | unexpected fluctuations in planned spend. |
• | changes to project timelines. |
• | the data generated from our research and development programs; |
• | our future operating performance, prospects and business strategy; |
• | the material risks related to our business and industry; |
• | the lack of an active public market for our ordinary and convertible preferred shares; |
• | the market performance of publicly traded companies in the life science and biotechnology sectors; |
• | the prices at which we issued ordinary and preferred shares and the superior rights and preferences of the preferred shares relative to the ordinary shares at the time of each grant; and |
• | the likelihood of achieving a liquidity events for the holders of our ordinary shares, series A preferred and series B preferred shares and G shares, such as an initial public offering, given prevailing market conditions. |
• | Tebentafusp, our ImmTAC molecule targeting an HLA-A*02:01 gp100 antigen, demonstrated monotherapy activity and recently achieved the primary endpoint of superior overall survival at the first pre-planned interim analysis of a randomized Phase 3 clinical trial in patients with previously untreated metastatic uveal melanoma. We anticipate submitting a BLA to the FDA in the third quarter of 2021, followed by a Marketing Authorization Application, or MAA, submission to the European Medicines Agency, or EMA. |
• | IMC-C103C, our ImmTAC molecule targeting an HLA-A*02:01 MAGE-A4 antigen, is currently being evaluated in a first-in-human, Phase 1/2 dose escalation trial in patients with solid tumor cancers including non-small-cell lung cancer, or NSCLC, gastric, head and neck, ovarian and synovial sarcoma. We believe this trial will demonstrate clinical activity of IMC-C103C, and we anticipate reporting Phase 1 initial data from this trial in the second half of 2021. We are developing this program under a co-development collaboration with Genentech, Inc., or Genentech, under which we have an option to retain 50% of the economics. |
• | IMC-F106C, our ImmTAC molecule targeting an optimal HLA-A*02:01 PRAME antigen identified with our MassSpec technology, is currently being evaluated in a first-in-human, Phase 1/2 dose escalation trial in patients with multiple solid tumor cancers including breast, endometrial, ovarian and small cell lung cancer, or SCLC. We believe this trial will demonstrate clinical activity of IMC-F106C, and we anticipate reporting Phase 1 initial data from this trial in mid-2022. |
• | GSK01, our ImmTAC molecule targeting an NY-ESO HLA-A*02:01 antigen, is currently being evaluated in the dose escalation phase of a Phase 1 clinical trial. When an optimal dosing regimen has been identified, a small expansion cohort of synovial sarcoma patients will be recruited to evaluate the clinical benefit of the therapeutic. This program is being developed under a collaboration with GlaxoSmithKline Intellectual Property Development Ltd, or GSK, which has an option to acquire full commercialization and development rights to this product candidate at the end of the ongoing Phase 1 clinical trial. |
• | IMC-I109V, our ImmTAV molecule targeting a conserved HBV envelope antigen, is our most advanced ImmTAV program and is currently being evaluated in a Phase 1/2 clinical trial in patients with chronic HBV who are non-cirrhotic, hepatitis B e-Antigen negative, and virally suppressed on chronic nucleot(s)ide analogue therapy. Our goal is to develop a functional cure for HBV and we anticipate commencing dosing in our Phase 1 single ascending dose, or SAD, trial in mid-2021. We are also developing a next-generation version of this molecule leveraging our research into universal HLA-E molecules which could benefit a much larger patient population as compared to classical-HLA antigens. |
• | IMC-M113V, our ImmTAV molecule targeting an HIV gag antigen bispecific TCR molecule, is currently in pre-clinical development. Our HIV programs are funded by the Bill & Melinda Gates Foundation, or the Gates Foundation, and we are required to make any successfully approved products available at reduced prices in certain developing countries. We retain full development and commercial right in non-developing countries. |
• | Secure marketing approval for, and then commercialize, tebentafusp, our lead ImmTAC, for the treatment of metastatic uveal melanoma. Our most advanced oncology therapeutic candidate, tebentafusp, has demonstrated superior overall survival benefit as a monotherapy in a randomized Phase 3 clinical trial in previously untreated metastatic uveal melanoma, a cancer that has historically proven to be insensitive to other immunotherapies. This primary endpoint was achieved with a hazard ratio of 0.51 (95% CI: 0.36, 0.71; p< 0.0001) at the first pre-planned interim analysis. We intend to seek regulatory approval for tebentafusp in the United States and Europe. We believe achieving regulatory approval of tebentafusp would provide validation of our entire ImmTAX platform. If tebentafusp is approved, we also believe it will present us with an attractive commercial opportunity, which we intend to pursue using a targeted commercialization strategy that requires minimal internal infrastructure. |
• | Advance our IMC-C103C program targeting MAGE-A4 for the treatment of solid tumors in collaboration with Genentech. We believe IMC-C103C has the potential to treat a wide range of solid tumors, including NSCLC. We are currently evaluating IMC-C103C in a first-in-human, Phase 1/2 dose escalation trial in patients with solid tumor cancers. We believe this trial will demonstrate clinical activity of IMC-C103C, and we anticipate reporting Phase 1 initial data from this trial in the second half of 2021. We are developing this program under a co-development collaboration with Genentech, and are jointly progressing clinical development of IMC-C103C with a partner who possesses deep expertise in clinical development and regulatory strategy. |
• | Advance our IMC-F106C program targeting PRAME for the treatment of solid tumors. IMC-F106C represents a significant commercial opportunity given the prevalence of the PRAME target across various cancers. PRAME is overexpressed in many solid tumors, including NSCLC, SCLC, endometrial, ovarian, esophageal, head and neck squamous cell carcinoma, and urothelial cancers. PRAME is also overexpressed in some hematological malignancies, including acute myeloid leukemia. PRAME expression is generally identified as a poor prognostic feature. We are currently evaluating IMC-F106C in a first-in-human, Phase 1/2 dose escalation trial in patients with solid tumor cancers including NSCLC, gastric, head and neck, ovarian and synovial sarcoma. We believe this trial will demonstrate clinical activity of IMC-F106C, and we anticipate reporting Phase 1 initial data from this trial in mid-2022. |
• | Advance our IMC-I109V program for the treatment of chronic HBV. Current standard-of-care antiviral agents for HBV do not provide a permanent cure in most cases. Therefore, lifelong treatment is necessary to lower the risk of chronic HBV-related complications and there remains a large unmet need for a functional cure. The goal of our IMC-I109V program is to develop a functional cure for chronic HBV. If successful, we believe our therapeutic will allow patients to have a finite period of |
• | Continue to develop our novel universal ImmTAX platform to meaningfully broaden the eligible patient pool. We are developing universal TCR therapeutics that are designed to be unrestricted by classical HLA status, which would have the potential to significantly increase the patient pool eligible for our therapeutics. Having pioneered the engineering of TCR bispecifics against classical HLA targets, we believe we are now at the forefront of ushering in a new era of TCR therapies by unlocking universal HLAs, such as HLA-E. This new approach, which we have validated in pre-clinical studies, offers the potential for all patients globally to benefit from a single therapeutic per target rather than requiring several classical HLA programs with their associated development costs. |
• | Continue to invest in our platform to discover and develop novel therapeutics. To remain an industry leader in TCR bispecifics, we intend to continue identifying and validating unique targets as well as optimizing current TCRs to continue to improve outcomes for patients across a broad range of diseases. |
• | Opportunistically pursue strategic partnerships to maximize the full potential of our pipeline and ImmTAX platform. We intend to selectively evaluate partnerships to explore combination therapies and access our partners’ industry-leading capabilities. We plan to assess opportunities to partner with large pharmaceutical companies in the areas of infectious disease and autoimmune diseases to access a broad commercial infrastructure for those indications. |
1. | Antibody-based Therapeutics – Engineered proteins derived from antibodies are able to recognize and bind cell surface antigens on both tumor and infected cells or modulate regulatory proteins, such as checkpoints, on the surface of immune cells. There are several types/classes of antibody-based therapeutics leveraging the properties of antibodies to treat disease, including for example: |
○ | Checkpoint Inhibitors – Checkpoint inhibition is an approach by which an antibody, called a checkpoint inhibitor, binds and blocks receptors on immune cells that function as negative regulators of the cell, which results in stimulation of T cell function and activation of an immune response. This approach is known as “releasing the break” on T cells and has been successfully employed in oncology where tumor cells often exploit these checkpoint molecules to turn off the immune response. |
○ | Bispecific T cell Engagers – These therapeutics are engineered antibodies able to recognize two cell surface targets, as opposed to one as is the case for a simple antibody, and redirect T cells to recognize and kill cancer by forming a bridge between the cancer cell and T cell. |
2. | Cell Therapies – Immune cells, often derived from the patient, engineered to be able to identify and target a specific antigen and disease. T cells are, in particular, the killer arm of the immune system. Because they can scan the body to identify abnormal or infected cells and only be activated once such identification is triggered, they play a critical role in the elimination of infections and tumors. This approach includes, among other therapeutics: |
○ | Antibody-Targeted CAR-T – These therapeutics are T cells extracted from a patient and engineered to express a chimeric antigen receptor, or CAR, on the cell surface. CARs are receptors which have the same binding properties as antibodies and are derived from the same molecular structure. These engineered T cells thus utilize antibody recognition to identify and target certain surface proteins/antigens on cancer cells. Upon binding, the T cell activation is triggered and can result/results in killing of the recognized cell. |
○ | T Cell Receptor (TCR) T cells – T cells extracted from a patient and engineered to express enhanced TCR molecules. TCRs, unlike antibodies, recognize a protein fragment or antigen presented on the cell surface in conjunction with a HLA complex. These TCRs can be engineered to recognize a specific cellular target associated with a tumor or infection. Upon binding, similarly to the CAR-T approach, the T cell will be activated and able to attack the cell. |
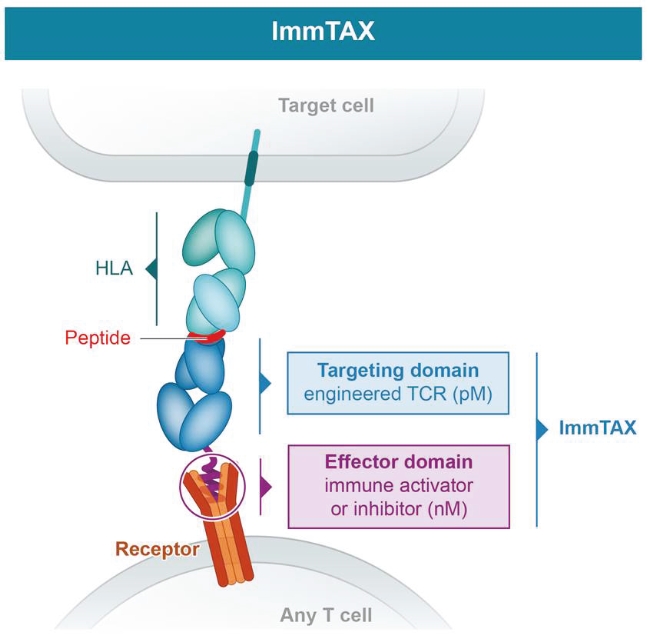
• | ImmTAC - Immune mobilizing monoclonal TCRs Against Cancer |
• | ImmTAV - Immune mobilizing monoclonal TCRs Against Viruses |
• | ImmTAAI - Immune modulating monoclonal TCRs Against AutoImmune disease |
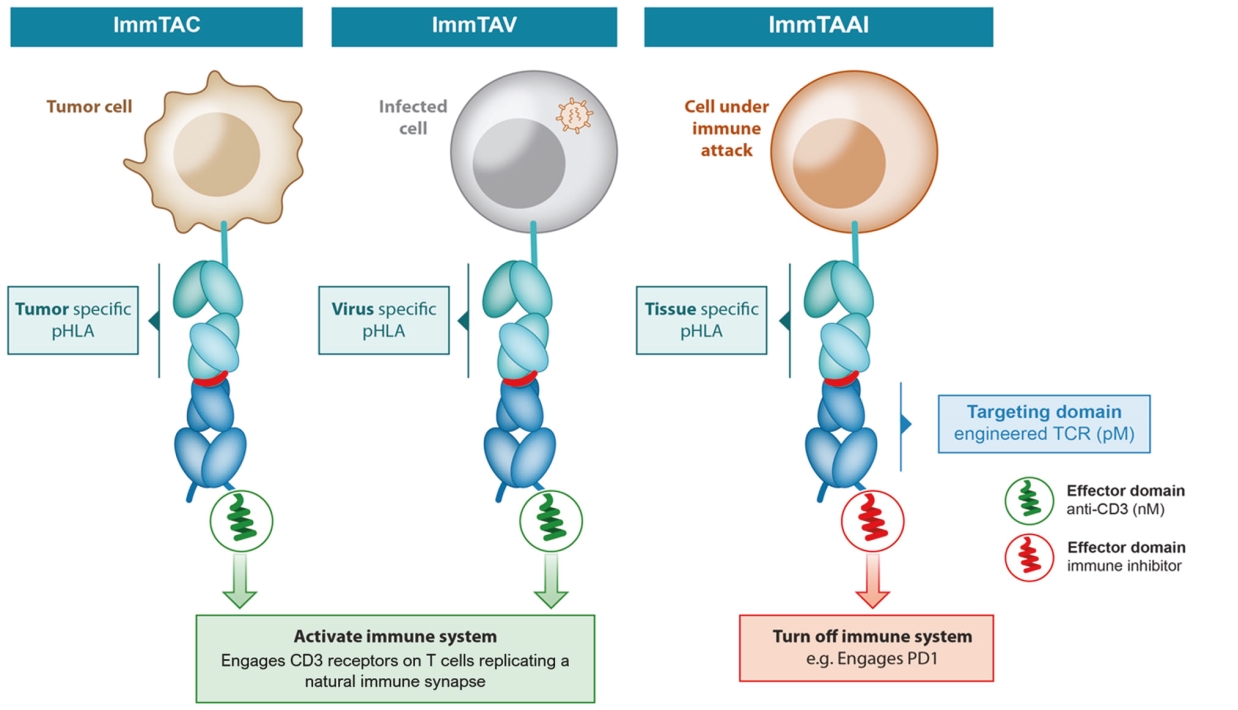
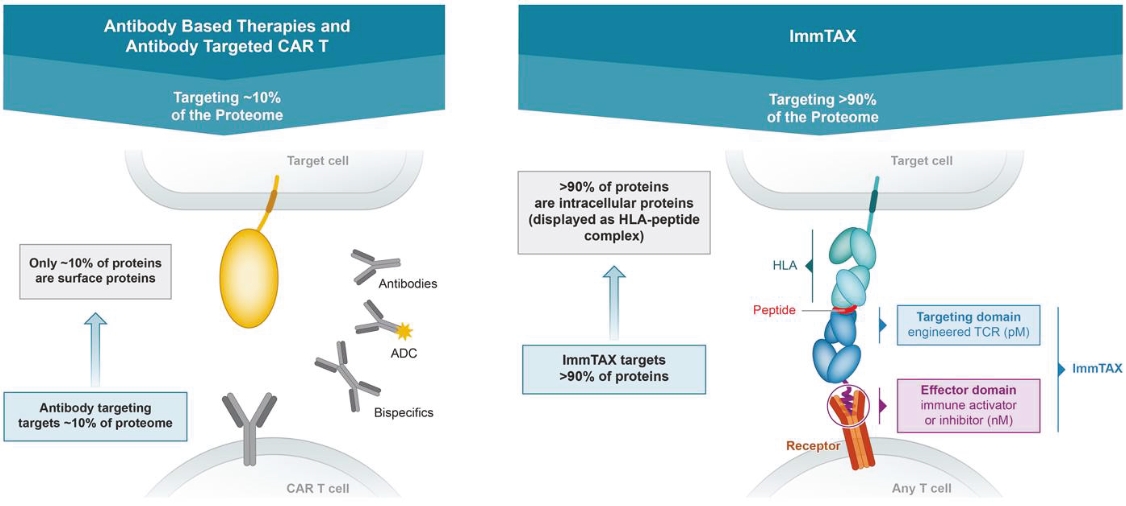
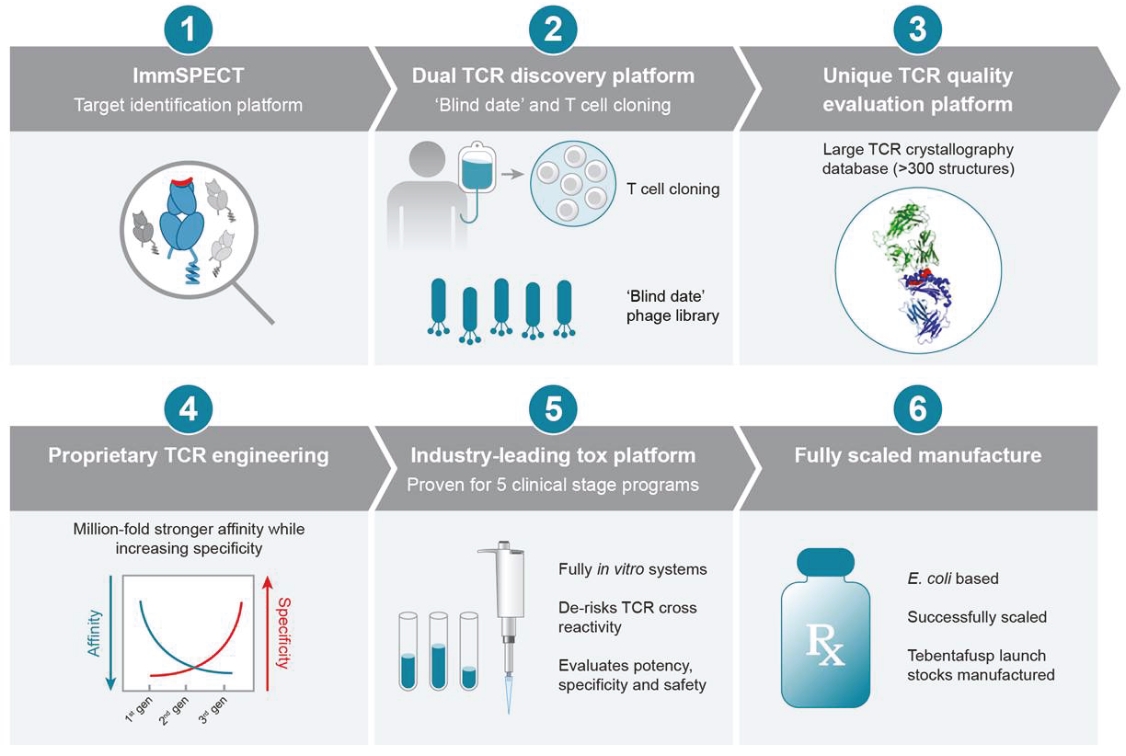
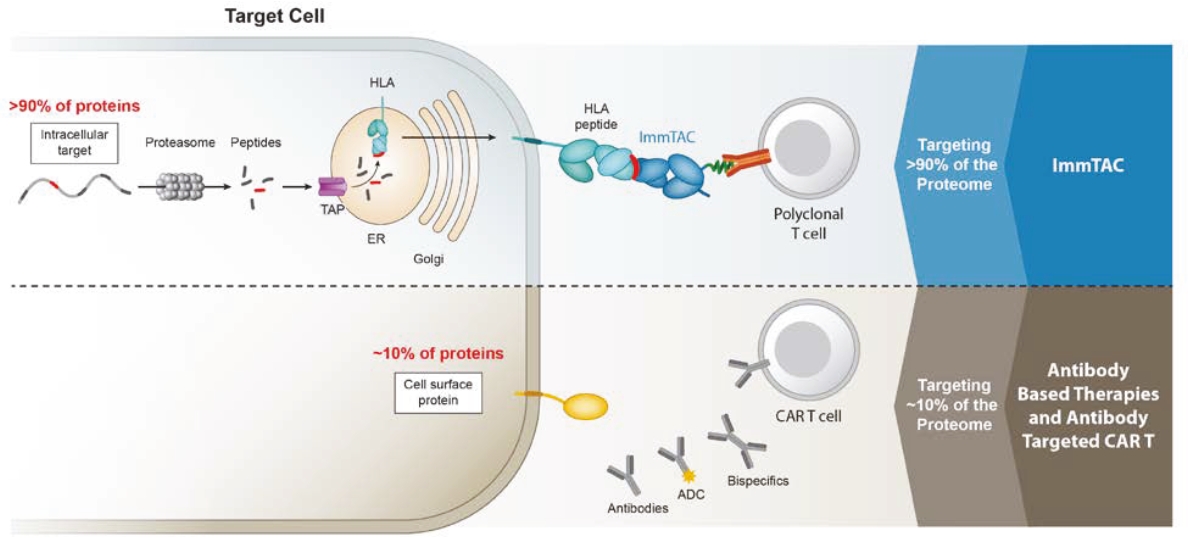
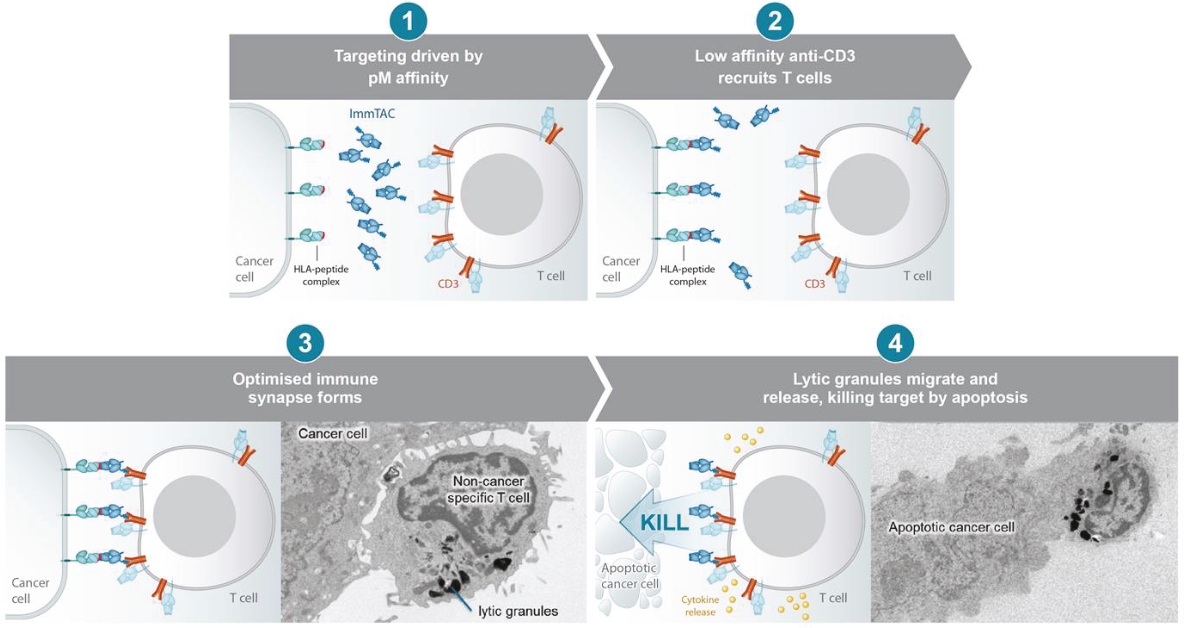
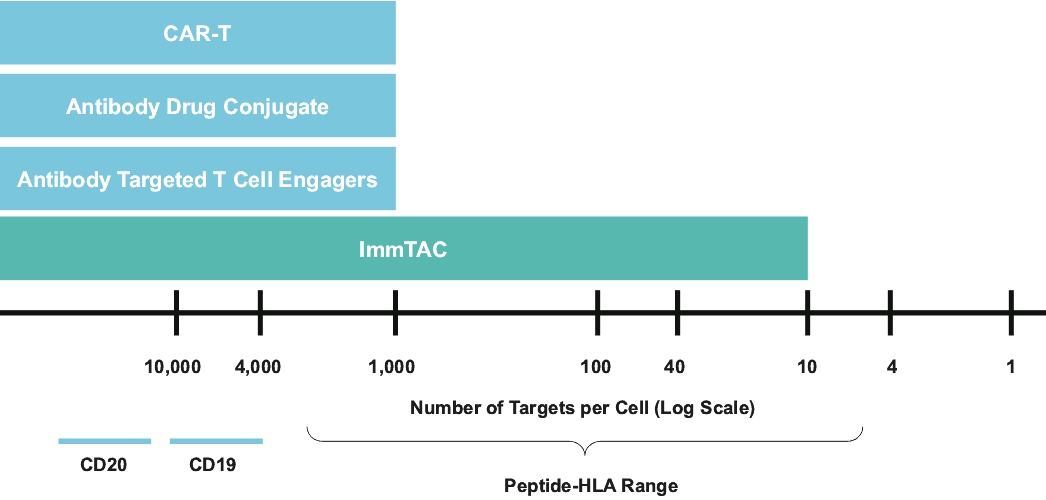
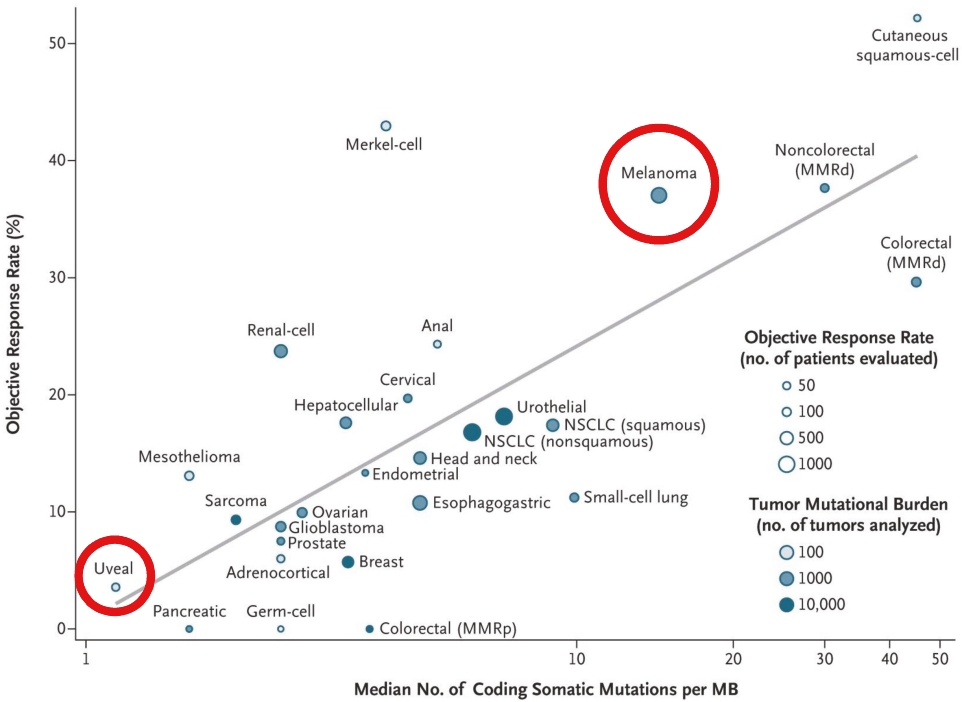
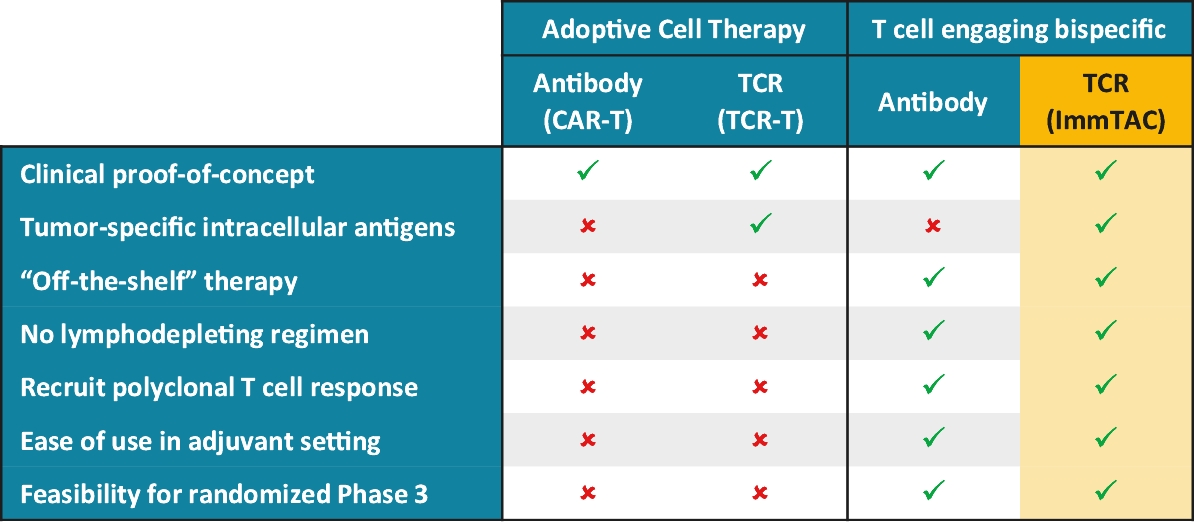
• | Phase 1 first-in-human clinical trial (n=84) demonstrated monotherapy activity per RECIST and immune related responses in uveal and cutaneous melanoma patients. |
• | Phase 2 clinical trial (n=127) demonstrated improved overall survival in a cross-trial comparison to a recent metanalysis based on prior clinical trials in a similar previously treated uveal melanoma patient population (n=287). The cross trial overall survival hazard ratio was 0.50 (95% CI 0.38,0.66). |
• | Phase 3 randomized clinical trial (n=378) achieved the primary endpoint of superior overall survival in the intent-to-treat population with a hazard ratio of 0.51 (95% CI: 0.36, 0.71), p<0.0001 favoring tebentafusp over investigators choice. |
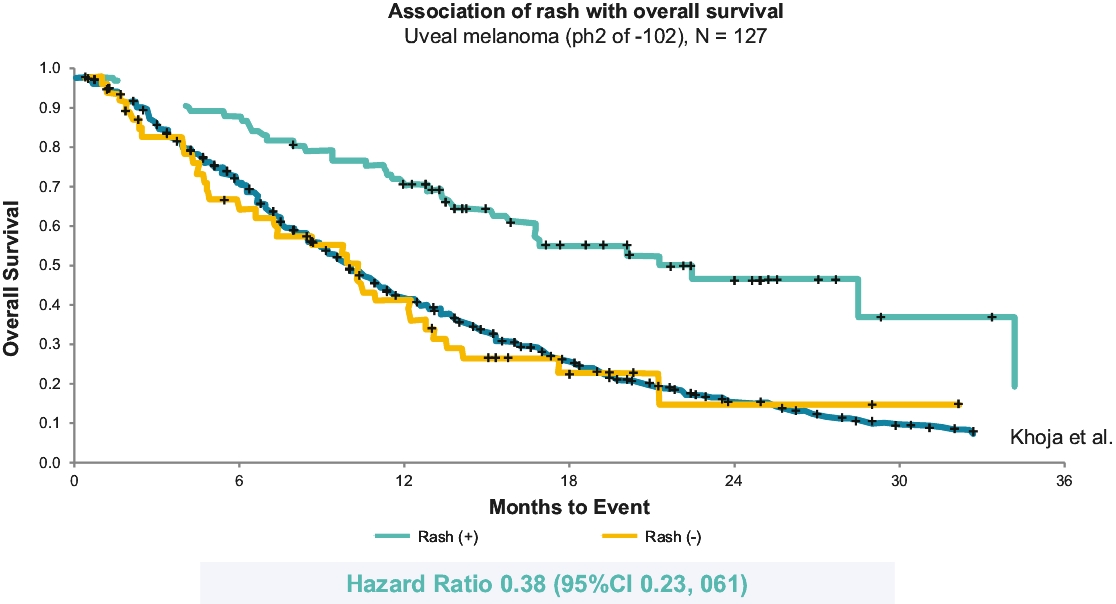
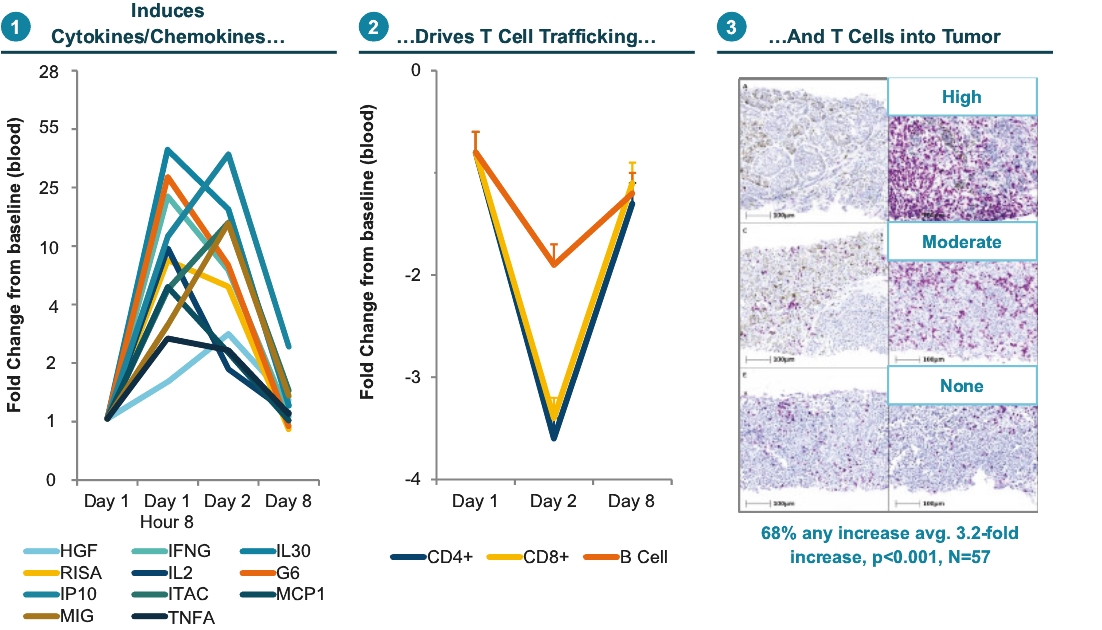
• | Phase 1 portion (dose escalation): This portion of the clinical trial defined the intra-patient dose escalation regimen, with a top dose of 68 mcg, which was then advanced as the recommended dose in the Phase 2 portion of the trial and our ongoing Phase 3 clinical trial. Of the 19 patients in the Phase 1 portion, we observed three patients had tumor responses that met the criteria defined by RECIST. An additional four patients did not meet RECIST criteria but had immune-related responses, a category of response previously described for the immune checkpoint therapies, and also suggested improved survival results that supported further studies. |
• | Phase 2 portion (expansion): This portion of the clinical trial was to evaluate the efficacy of tebentafusp in 127 patients with metastatic uveal melanoma as a second-line or later treatment. The primary endpoint was to estimate the objective response rate, or ORR, under RECIST 1.1 according to an independent central review committee. We believe the observation of immune related responses in the Phase 1 portion of this trial and the Phase 1 first-in-human trial indicates that overall survival, which captures benefit from RECIST and immune related responses, is a better measurement of treatment effect for tebentafusp, and thus was included as a secondary endpoint of this trial. Of the 127 metastatic uveal melanoma patients treated, all had received prior treatments and the majority had received prior immunotherapy regimens (73.2% had prior immunotherapy; 65.4% had prior anti-PD-1). |
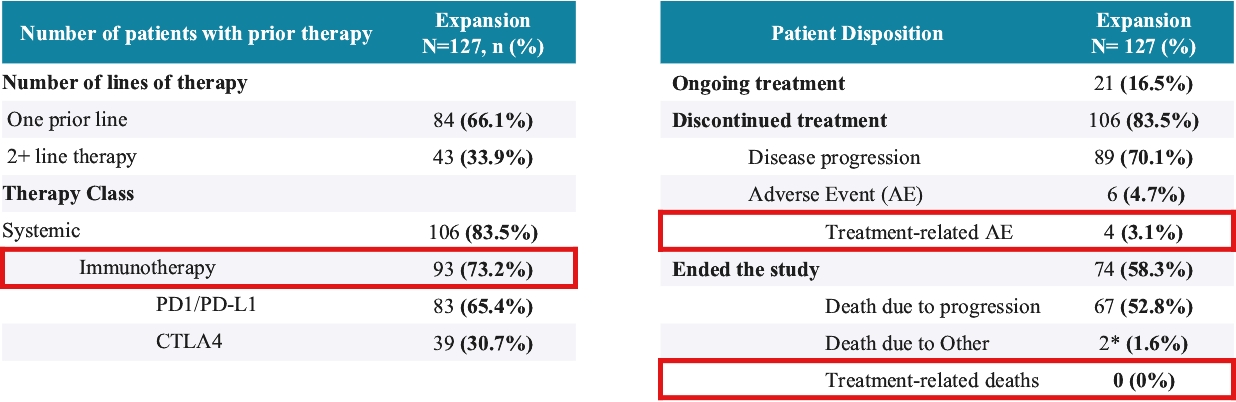
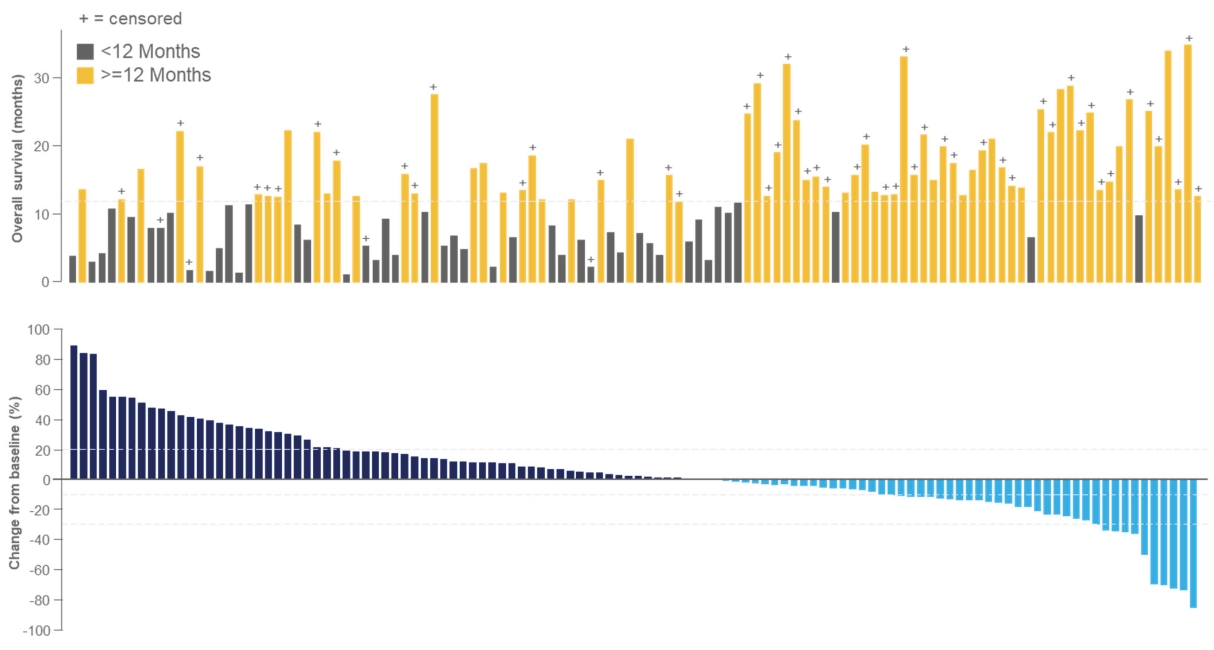
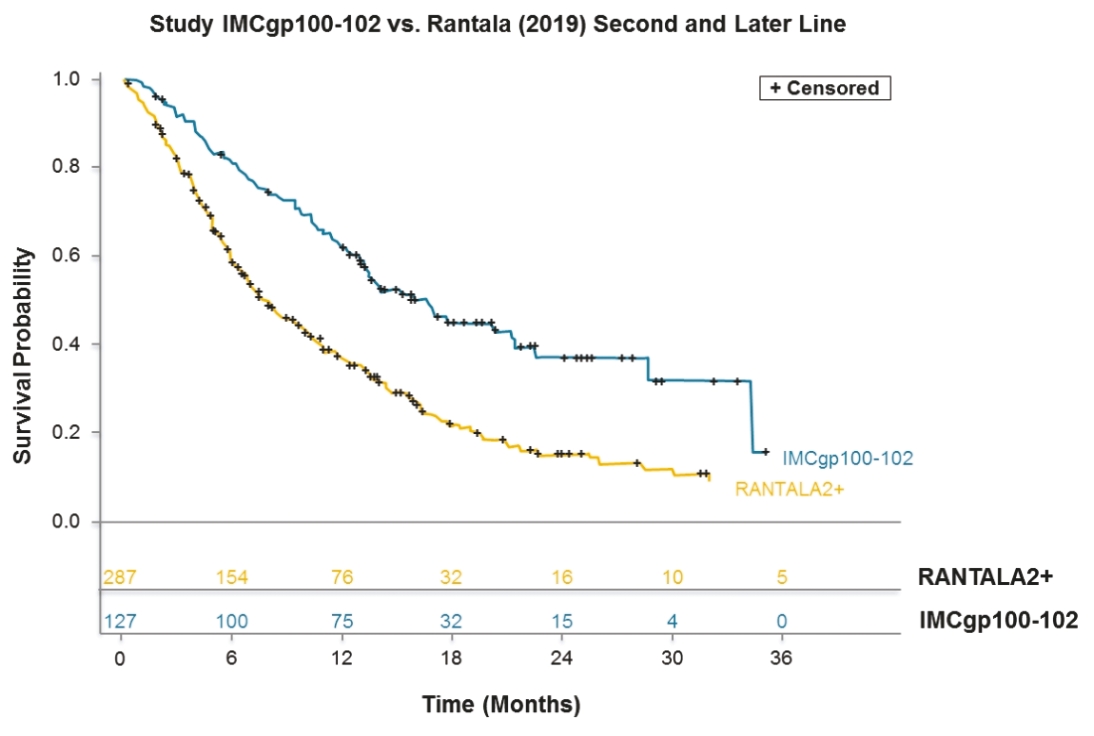
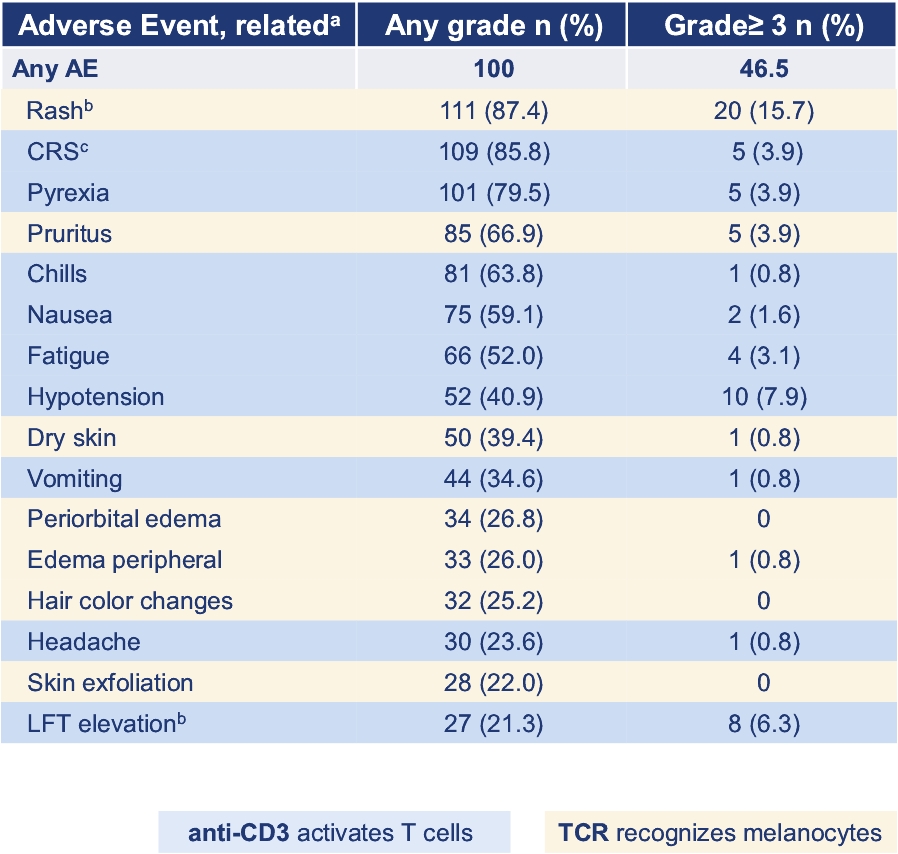
a. | Investigator assignment of causality; |
b. | LFT elevation and rash are composites of preferred terms; |
c. | CRS assesed retrospectively by ASTCT (Lee 2019) criteria |
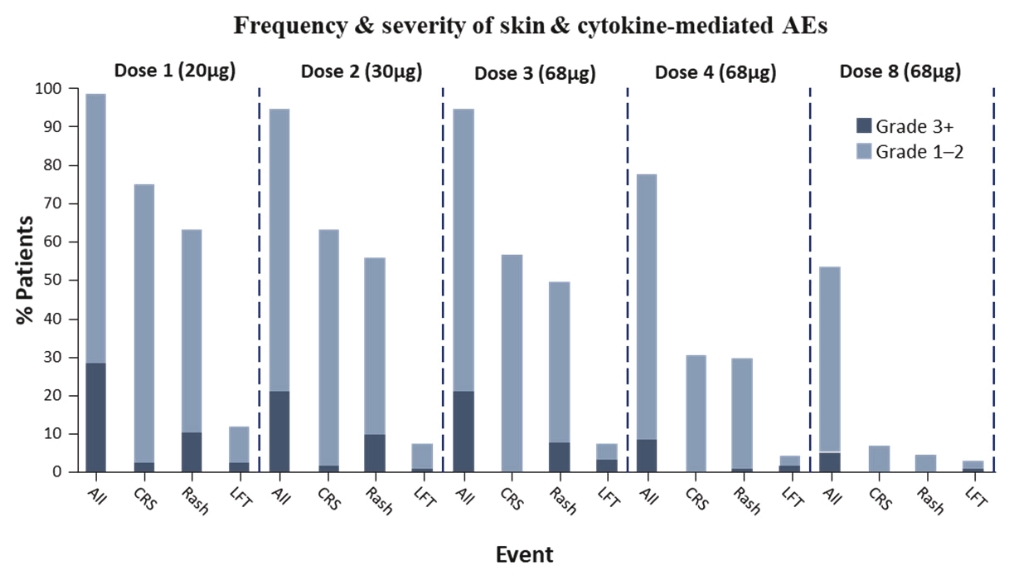
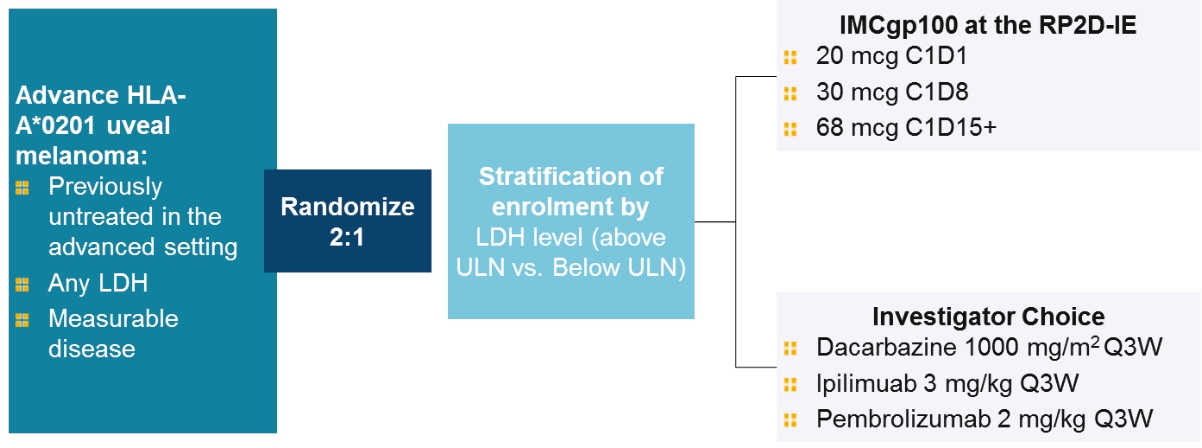
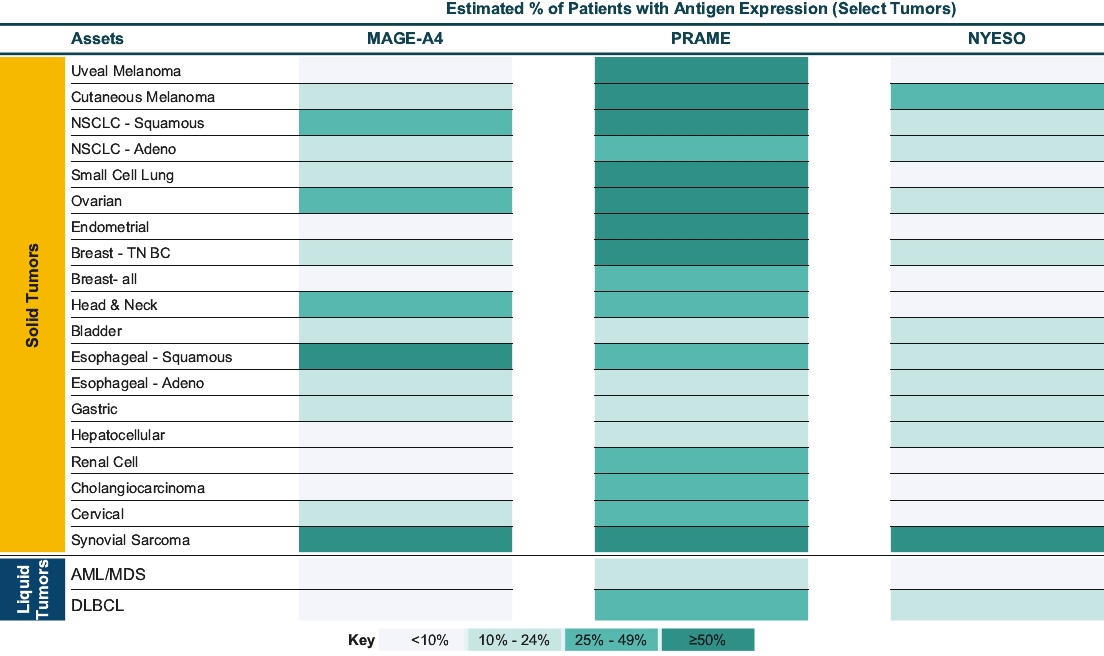
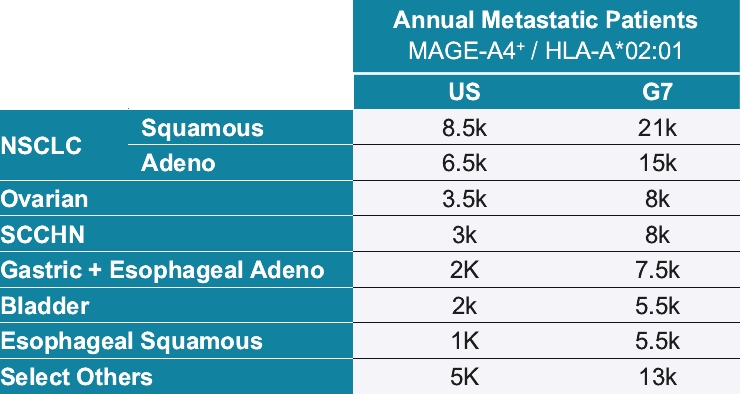
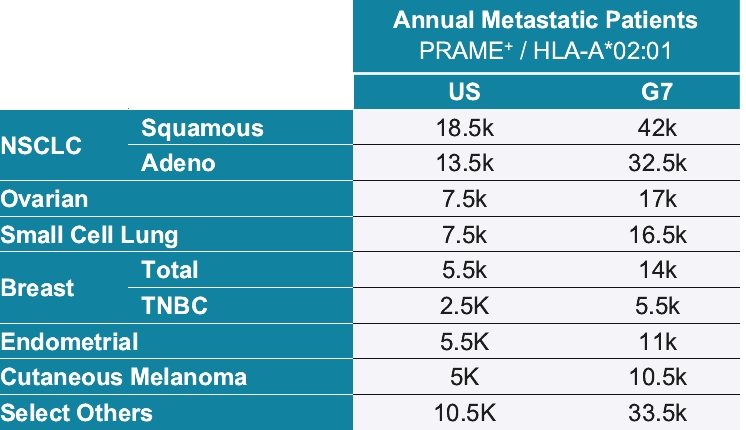
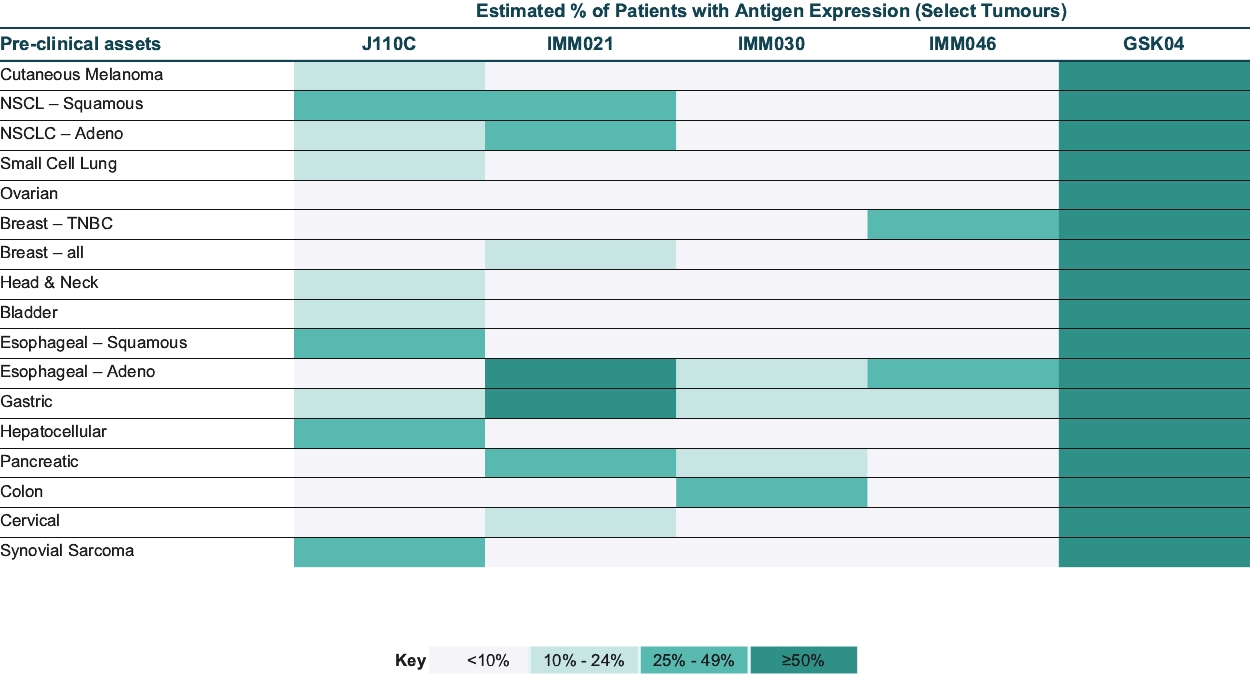
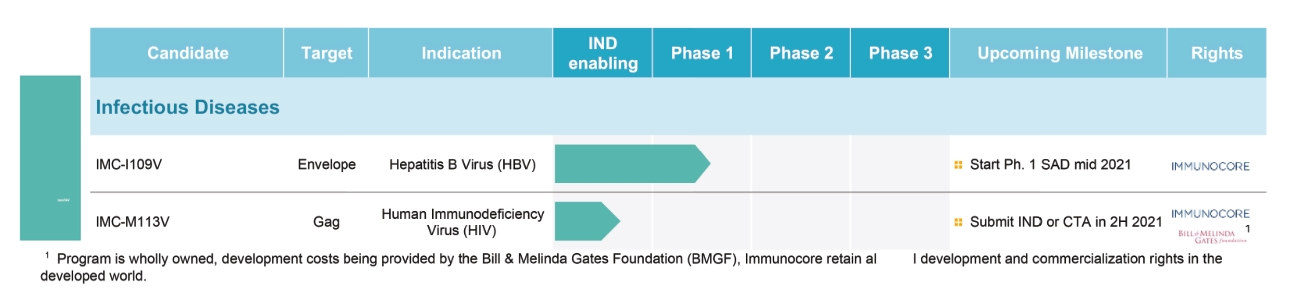
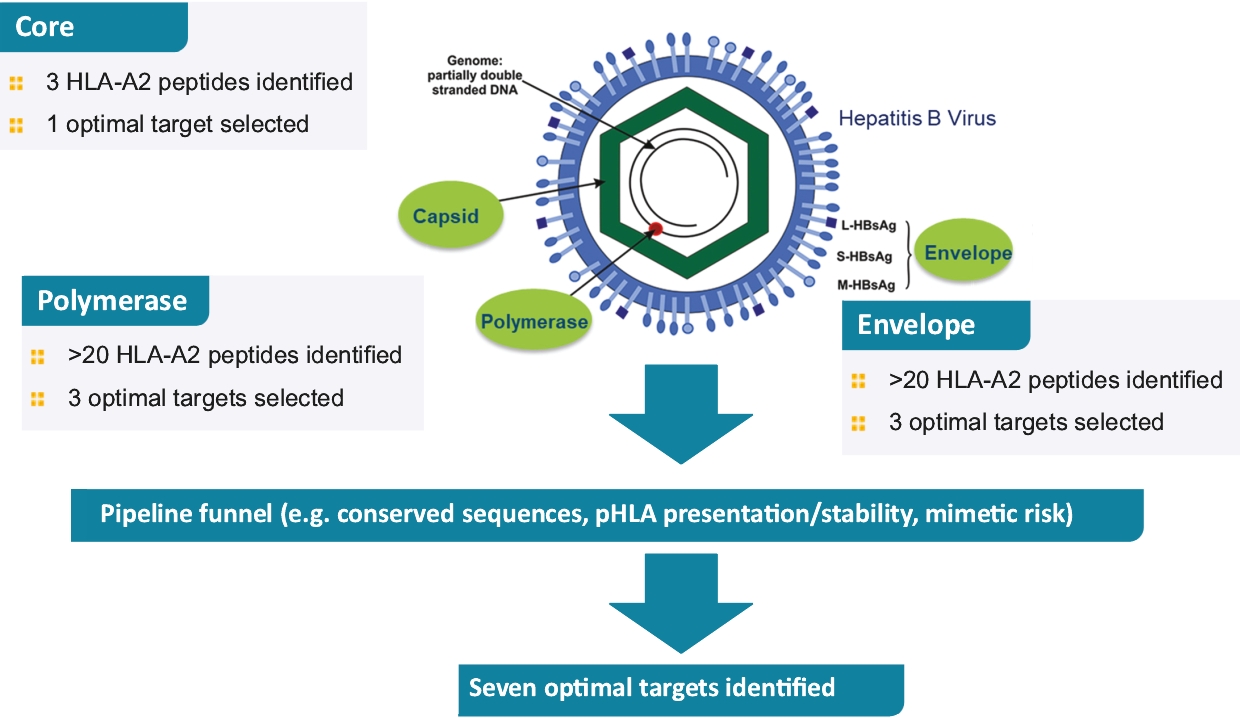
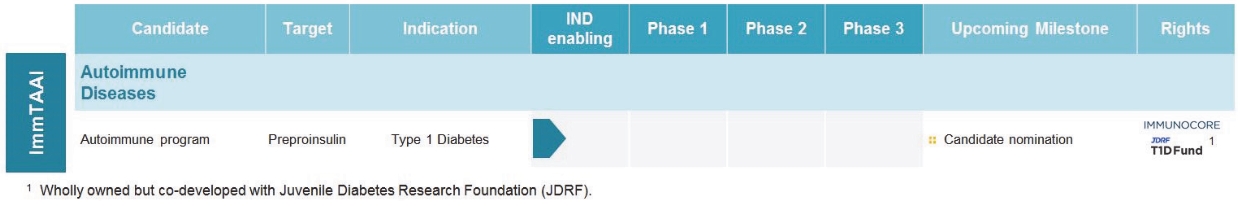
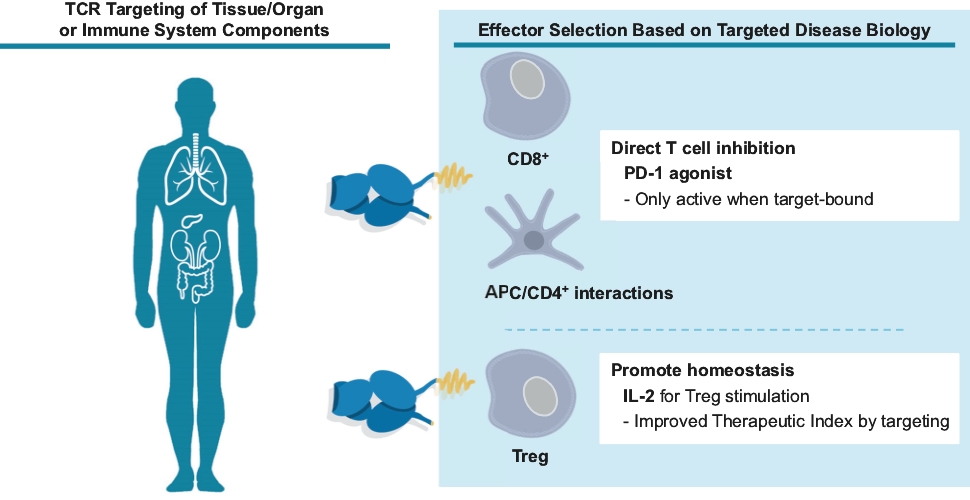
• | HLA-E target identification and validation: HLA-E peptide antigens are significantly more unstable than classical HLA peptides and fall apart within minutes rather than hours. Therefore, we have developed a suite of four new HLA-E target identification and validation assays that have allowed us to identify novel HLA-E targets for HBV, HIV, TB and a number of oncology targets. |
• | Antigen stabilization: HLA-E/peptide instability also makes the isolation and engineering of specific TCRs challenging. We developed and patented a new HLA-E stabilization approach that allows highly specific TCRs to be isolated and engineered. |
• | Sufficiently high specificity: HLA-E presents peptides that tend to have a high degree of similarity in their sequence, making it challenging to introduce sufficient levels of specificity to support clinical development. We have successfully adapted existing specificity tools to overcome these challenges. |
• | Biovian Ltd., headquartered in Turku, Finland, for early-phase clinical drug substance and drug product cGMP manufacturing; |
• | AGC Biologics A/S, headquartered in Copenhagen, Denmark, for late-phase clinical and commercial scale drug substance cGMP manufacturing; and |
• | Baxter Oncology GmbH, headquartered in Halle/Westfalen, Germany for late-phase clinical and commercial scale drug product cGMP manufacturing. |
• | nonclinical laboratory tests, animal studies and formulation studies all performed in accordance with the FDA’s good laboratory practices, or GLP, regulations; |
• | submission to the FDA of an IND application for human clinical testing, which must become effective before human clinical trials may begin; |
• | approval by an institutional review board, or IRB, representing each clinical site before each clinical trial may be initiated; |
• | performance of adequate and well-controlled human clinical trials to establish the safety, potency and purity of the product candidate for each proposed indication, in accordance with Good Clinical Practices, or GCP; |
• | preparation and submission to the FDA of a BLA for a biological product requesting marketing for one or more proposed indications, including submission of detailed information on the manufacture and composition of the product in clinical development and proposed labeling; |
• | review of the product by an FDA advisory committee, if applicable; |
• | one or more FDA inspections of the manufacturing facility or facilities, including those of third parties, at which the product, or components thereof, are produced to assess compliance with current Good Manufacturing Practices, or cGMP, requirements and to assure that the facilities, methods and controls are adequate to preserve the product’s identity, strength, quality and purity; |
• | FDA audits of the clinical study sites to assure compliance with GCPs, and the integrity of clinical data in support of the BLA; |
• | payment of user fees and securing FDA approval of the BLA and licensure of the new biological product; and |
• | compliance with any post-approval requirements, including the potential requirement to implement a Risk Evaluation and Mitigation Strategy, or REMS, and any post-approval studies required by the FDA. |
• | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
• | fines, untitled letters or warning letters or holds on post-approval clinical trials; |
• | refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of product license approvals; |
• | product seizure or detention, or refusal to permit the import or export of products; or |
• | injunctions or the imposition of civil or criminal penalties. |
• | the federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in cash or kind, in exchange for, or to induce, either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made, in whole or in part, under federal healthcare programs such as the Medicare and Medicaid programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers, on the one hand, and prescribers, purchasers, formulary managers and other individuals and entities on the other. The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the ACA, amended the intent requirement of the federal Anti-Kickback Statute such that a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it in order to commit a violation; |
• | the federal civil and criminal false claims, including the civil False Claims Act, or the FCA, and civil monetary penalties laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid or other third-party payors that are false or fraudulent, or knowingly making, or causing to be made, a false record or statement material to a false or fraudulent claim to avoid, decrease, or conceal an obligation to pay money to the federal government. Certain marketing practices, including off-label promotion, also may implicate the FCA. In addition, the ACA codified case law that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the FCA. |
• | HIPAA imposes criminal and civil liability, among other things, for executing, or attempting to execute, a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; |
• | the federal Physician Payments Sunshine Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program, with specific exceptions, to report annually to the Centers for Medicare & Medicaid Services, or CMS, information related to payments and other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, and ownership and investment interests held by physicians and their immediate family members. Effective January 1, 2022, applicable manufacturers will also be required to report such information regarding its relationships with physician assistants, nurse practitioners, clinical nurse specialists, anesthesiologist assistants, certified registered nurse anesthetists and certified nurse midwives during the previous year; |
• | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, and their implementing regulations, which imposes certain obligations, including mandatory contractual terms, with respect to safeguarding the transmission, security and privacy of individually identifiable health information on covered entities, such as health plans, health care clearinghouses and certain healthcare providers, and their respective business associates that create, receive, maintain or transmit individually identifiable health information for or on behalf of a covered entity, and their subcontractors that use, disclose, access, or otherwise process individually identifiable protected health information; and |
• | state and foreign law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government that otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers, drug pricing and/or marketing expenditures; state and local laws requiring the registration of |
• | an annual, nondeductible fee on any entity that manufactures or imports specified branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs; |
• | an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program to 23.1% and 13.0% of the average manufacturer price for most branded and generic drugs, respectively; |
• | a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected; |
• | extension of a manufacturer’s Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; |
• | expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to certain individuals with income at or below 133% of the federal poverty level, thereby potentially increasing a manufacturer’s Medicaid rebate liability; |
• | a new Medicare Part D coverage gap discount program, in which manufacturers must now agree to offer 70% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for a manufacturer’s outpatient drugs to be covered under Medicare Part D; |
• | expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program; and |
• | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. |
| | | At December 31, | |||||||
| | | 2018 | | | 2019 | | | 2020 | |
Function: | | | | | | | |||
Administrative | | | 67 | | | 67 | | | 55 |
Research and development | | | 394 | | | 392 | | | 236 |
Total | | | 461 | | | 459 | | | 291 |
| | | | | | | ||||
Geography: | | | | | | | |||
United Kingdom | | | 422 | | | 409 | | | 242 |
European Union | | | 2 | | | 3 | | | 2 |
United States | | | 37 | | | 47 | | | 47 |
Name | | | Age | | | Position(s) |
Executive Officers: | | | | | ||
Bahija Jallal, Ph.D. | | | 59 | | | Chief Executive Officer and Director |
Brian Di Donato | | | 54 | | | Chief Financial Officer and Head of Strategy |
David Berman, M.D., Ph.D. | | | 50 | | | Head of Research and Development |
Non-Executive Directors: | | | | | ||
Professor Sir John Bell | | | 68 | | | Chairman of the Board of Directors |
Travis Coy | | | 40 | | | Director |
Roy S. Herbst, M.D., Ph.D | | | 58 | | | Director |
Robert Perez | | | 56 | | | Director |
Kristine Peterson | | | 61 | | | Director |
Professor Sir Peter Ratcliffe | | | 66 | | | Director |
• | Exemption from filing quarterly reports on Form 10-Q containing unaudited financial and other specified information or current reports on Form 8-K upon the occurrence of specified significant events; |
• | Exemption from Section 16 rules requiring insiders to file public reports of their securities ownership and trading activities and providing for liability for insiders who profit from trades in a short period of time; |
• | Exemption from quorum requirements for shareholder meetings. In accordance with usual practice in England and Wales, our articles of association will provide alternative quorum requirements that are generally applicable to shareholder meetings; |
• | Exemption from the Nasdaq rules applicable to domestic issuers requiring disclosure within four business days of any determination to grant a waiver of the code of business conduct and ethics to directors and officers; |
• | Exemption from the requirement to obtain shareholder approval for certain issuances of securities, including shareholder approval of share option plans; |
• | Exemption from the requirement that our audit committee have review and oversight responsibilities over all “related party transactions,” as defined in Item 7.B of Form 20-F; |
• | Exemption from the requirement that our board have a compensation committee that is composed entirely of independent directors with a written charter addressing the committee’s purpose and responsibilities; and |
• | Exemption from the requirements that director nominees are selected, or recommended for selection by our board, either by (1) independent directors constituting a majority of our board’s independent directors in a vote in which only independent directors participate, or (2) a committee comprised solely of independent directors, and that a formal written charter or board resolution, as applicable, addressing the nominations process is adopted. |
• | determining whether to appoint, reappoint or remove any auditors, and making recommendations to the board of directors to be put to the shareholders for approval at the annual general meeting; |
• | reviewing audit plans, the adequacy of staffing and fees, whilst overseeing the negotiation and execution of any engagement letters on behalf of the Company; |
• | at least annually, assessing the qualifications, performance, and independence of the auditors, or in the case of prospective auditors, before they are engaged; |
• | overseeing the policies and procedures governing how the Company may employ individuals who are or once were employed by the auditors; |
• | reviewing results of the annual audit, audited financial statements, periodic and annual reports, earnings announcements, proxy report, accounting principles and policies; |
• | evaluating management’s cooperation with the auditors during their audit examination; |
• | reviewing and reporting on policies on financial risk management and assessment; |
• | reviewing the audit plan of any internal audit team; |
• | reviewing the scope, design, adequacy and effectiveness of internal controls; |
• | reviewing correspondence with regulators or governmental agencies that raise material issues regarding the Company’s financial statements or accounting policies; |
• | overseeing procedures for receiving, retaining and investigating complaints; |
• | monitoring compliance with company’s Code of Business Conduct and Ethics and related party transactions rules; and |
• | reviewing with management legal and regulatory compliance and any actual, pending, or threatened legal or financial matters that could significantly affect the Company’s business or financial statements or as otherwise deemed appropriate by the audit committee. |
• | reviewing, modifying and overseeing the company’s overall compensation strategy and policies; |
• | reviewing and approving the compensation and other terms of employment of the company’s Chief Executive Officer; |
• | reviewing and approving all elements of the compensation and other terms of employment of the executive officers and other senior management reporting directly to the Chief Executive Officer; |
• | reviewing and recommending to the board of directors for its approval the type and amount of compensation to be paid or awarded to members of the board of directors; |
• | undertaking sole responsibility for the appointment, authority to select, retain, and terminate any compensation and oversight of the work of compensation consultants, legal counsel, or any other advisors engaged for the purpose of advising the remuneration committee; |
• | exercising full power and authority to adopt, amend, terminate, and administer the Company’s equity award, pension, and profit sharing plans, incentive plans, bonus plans, executive benefit plans, stock purchase plans, deferred compensation plans and other similar programs; |
• | reviewing and discussing with management the company’s Compensation Discussion and Analysis section of the company’s annual reports, registration statements, proxy statements, or information statements filed with the SEC; |
• | reviewing and discussing with management any conflicts of interest raised; and |
• | overseeing the preparation of any report required by applicable U.S. and U.K. rules and regulations to be included in the company’s public filings relating to compensation policy and practices, including but not limited to the directors’ remuneration report required under the Companies Act. |
• | identifying and evaluating candidates, including nomination of incumbent directors for re-election and nominees recommended by shareholders to serve on the board of directors; |
• | making recommendations to the board of directors regarding nominees for directors at the next annual general meeting; |
• | periodically reviewing the performance of the board of directors, including committees of the board of directors and management; |
• | overseeing the board of directors' committee structure and operations, including authority to delegate to subcommittees and committee reporting to the board of directors; |
• | reviewing with the Chief Executive Officer the succession plans for the Company’s executive officers; |
• | instituting plans or programs for the continuing education of directors and orientation of new directors, as it deems appropriate; and |
• | periodically reviewing the processes and procedures to provide information to the board of directors and its committees. |
1. | Annual Board of Directors Service Retainer: |
a. | All Eligible Directors: $40,000 |
b. | Independent Chair of the Board of Directors Service Retainer (in addition to Eligible Director Service Retainer): $30,000 |
2. | Annual Committee Member Service Retainer: |
a. | Member of the Audit Committee: $7,500 |
b. | Member of the Remuneration Committee: $5,000 |
c. | Member of the Nominating and Corporate Governance Committee: $4,000 |
3. | Annual Committee Chair Service Retainer (in addition to Annual Committee Member Service Retainer): |
a. | Chair of the Audit Committee: $7,500 |
b. | Chair of the Remuneration Committee: $5,000 |
c. | Chair of the Nominating and Corporate Governance Committee: $4,000 |
• | an attempt to transfer, assign or encumber the option (save for a transfer to a personal representative on death); |
• | a performance condition failing to be met that results in the entire option being incapable of exercise; |
• | the date stated in the relevant option certificate; |
• | the first anniversary of an option holder’s death; |
• | 90 days after the option holder ceases to be employed by the company; |
• | if the board of directors uses its discretion to permit early exercise of an option within a defined period determined by the board of directors, the expiry of such period; |
• | 40 days after the completion of a Takeover or an Asset Sale (both as defined below) (or immediately after completion if option holders are given the opportunity to exercise their options by the board of directors prior to completion); |
• | 40 days after a reorganization of the company if a replacement option is offered in the acquirer as part of the reorganization; or |
• | the option holder becoming bankrupt. |
• | they are a Good Leaver, in which case they may exercise their vested option and 50% of their unvested option (calculated as at the date the option holder ceased to employed) for a period ending 90 days after becoming a Leaver, or 12 months from the date of death if the reason for leaving is due to an option holder’s death; or |
• | they are Bad Leaver, in which case they may exercise their vested option (calculated as at the date the option holder ceased to employed) for a period ending 90 days after becoming a Leaver; or |
• | the board of directors determines otherwise. |
• | an attempt to transfer, assign or encumber the option (save for a transfer to a personal representative on death); |
• | a performance condition failing to be met that results in the entire option being incapable of exercise; |
• | the date stated in the relevant option certificate; |
• | the first anniversary of an option holder’s death; |
• | 90 days after the option holder ceases to be employed or engaged by the company; |
• | if the board of directors uses its discretion to permit early exercise of an option within a defined period determined by the board of directors, the expiry of such period; |
• | 40 days after the completion of a Takeover or an Asset Sale (or immediately after completion if option holders are given the opportunity to exercise their options by the board of directors prior to completion); or |
• | the option holder becoming bankrupt. |
• | they are a Good Leaver, in which case they may exercise their vested option and 50% of their unvested option (calculated as at the date the option holder ceased to employed) for a period ending 90 days after becoming a Leaver, or 12 months from the date of death if the reason for leaving is due to an option holder’s death; or |
• | they are Bad Leaver, in which case they may exercise their vested option (calculated as at the date the option holder ceased to employed) for a period ending 90 days after becoming a Leaver; or |
• | the board of directors determines otherwise. |
Participants | | | Series C Preferred Shares (#) | | | Ordinary Shares (#) |
Entities affiliated with Baker Brothers | | | 274,574 | | | 4,965 |
Entities affiliated with General Atlantic | | | 219,659 | | | 18,963 |
Eli Lilly S.A. | | | — | | | 23,238 |
• | grants our preferred shareholders specified registration rights with respect to our shares held by them; |
• | obligates us to deliver periodic financial statements and other information to certain of the shareholders who are parties to the Series C Shareholders’ Agreement; and |
• | provides for certain appointment rights with respect to our board of directors and the voting of shares in favor of specified transactions approved by our board of directors and the requisite majority of our shareholders. |
Participants | | | Series B Perferred Shares (#) |
Entities affiliated with General Atlantic(1) | | | 555,893 |
Eli Lilly S.A. | | | 71,588 |
(1) | These shares were purchased by GA IMC Holding, L.P. |
• | contemplates granting our preferred shareholders specified registration rights with respect to our shares held by them, which is to be memorialized in a registration rights agreement that we intend to enter into prior to the completion of this offering; |
• | obligates us to deliver periodic financial statements to certain of the shareholders who are parties to the Series B Shareholders’ Agreement; and |
• | provides for certain appointment rights with respect to our board of directors and the voting of shares in favor of specified transactions approved by our board of directors and the requisite majority of our shareholders. |
• | each beneficial owner of 5% or more of our outstanding ordinary shares and non-voting ordinary shares; |
• | each of our directors and executive officers; and |
• | all of our directors and executive officers as a group. |
| | | Number of Ordinary Shares Beneficially Owned | | | Percentage of Ordinary Shares Beneficially Owned | ||||
Name of Beneficial Owner | | | Before Offering | | | After Offering | |||
5% or Greater Shareholders: | | | | | | | |||
Entities affiliated with General Atlantic(1) | | | 3,972,575 | | | 12.4% | | | 9.6% |
Eli Lilly S.A.(2). | | | 2,548,145 | | | 7.9% | | | 6.2% |
Nicholas John Cross(3) | | | 2,369,610 | | | 7.4% | | | 5.7% |
Ian Laing(4) | | | 2,364,420 | | | 7.4% | | | 5.7% |
Malin Life Sciences Holdings Limited(5) | | | 2,359,425 | | | 7.4% | | | 5.7% |
George Edward Silvanus Robinson(6) | | | 2,153,520 | | | 6.7% | | | 5.2% |
Blackwell Family(7) | | | 1,719,505 | | | 5.4% | | | 4.2% |
Entities affiliated with Baker Brothers(8) | | | 1,663,255 | | | 5.2% | | | 4.0% |
Schroders UK Public Private Trust plc(9) | | | 1,595,585 | | | 5.0% | | | 3.9% |
Executive Officers and Directors: | | | | | | | |||
Bahija Jallal, Ph.D. | | | — | | | — | | | — |
Brian Di Donato | | | — | | | — | | | — |
David Berman, M.D., Ph.D. | | | — | | | — | | | — |
Professor Sir John Bell(10) | | | 5,760 | | | * | | | * |
Travis Coy | | | — | | | — | | | — |
Roy Herbst | | | — | | | — | | | — |
Robert Perez | | | — | | | — | | | — |
Kristine Peterson | | | — | | | — | | | — |
| | | Number of Ordinary Shares Beneficially Owned | | | Percentage of Ordinary Shares Beneficially Owned | ||||
Name of Beneficial Owner | | | Before Offering | | | After Offering | |||
All current directors and executive officers as a group (8 persons)(11) | | | 5,760 | | | * | | | * |
* | Represents beneficial ownership of less than one percent. |
(1) | Consists of 3,972,575 ordinary shares issuable upon conversion of series B preferred shares, series C preferred shares and ordinary shares held by GA IMC Holding, L.P. The limited partners that share beneficial ownership of the shares held by GA IMC Holding are the following General Atlantic investment funds: General Atlantic Partners (Bermuda) EU, L.P. (“GAP EU”), General Atlantic Partners (Bermuda) IV, L.P. (“GAP IV”) , GAP Coinvestments III, LLC (“GAPCO III”), GAP Coinvestments IV, LLC (“GAPCO IV”), GAP Coinvestments V, LLC (“GAPCO V”) and GAP Coinvestments CDA, LLC (“GAPCO CDA”). The general partner of GAP EU and GAP IV is General Atlantic GenPar (Bermuda), L.P. (“GenPar Bermuda”). GAP (Bermuda) Limited (“GAP (Bermuda) Limited”) is the general partner of GenPar Bermuda. General Atlantic’s address is c/o Conyers Client Services (Bermuda) Limited, Clarendon House, 2 Church Street, Hamilton MM II, Bermuda. |
(2) | Consists of 2,548,145 ordinary shares issuable upon conversion of (a) 39,703 ordinary shares held by Eli Lilly S.A., (b) 398,338 series A preferred shares held by Eli Lilly S.A (c) 71,588 series B preferred shares and (d) 39,703 ordinary shares held by Eli Lilly S.A. Eli Lilly S.A.’s address is 16, Chemin des Coquelicots, 12 Geneva, Switzerland. |
(3) | Consists of 2,369,610 ordinary shares issuable upon conversion of (a) 467,458 ordinary shares held by Mr. Cross (b) 6,462 series A preferred shares and (c) 2 series B preferred shares held by Mr. Cross. |
(4) | Consists of (a) 1,915,140 ordinary shares issuable upon conversion of (i) 377,792 ordinary shares held by Mr. Laing, (ii) 5,234 series A preferred shares held by Mr. Laing, (iii) 2 series B preferred shares and (iv) 1,038 ordinary shares underlying options exercisable within 60 days of December 31, 2020 held by Mr. Laing and (b) 449,280 ordinary shares issuable upon conversion of (i) 88,628 ordinary shares held by Ms. Laing , (ii) 1,228 series A preferred shares held by Ms. Laing. |
(5) | Consists of 2,359,425 ordinary shares issuable upon conversion of (a) 46,991 ordinary shares held by Malin Life Sciences Holdings Limited and (b) 424,894 series A preferred shares held by Malin Life Sciences Holdings Limited. Malin Life Sciences Holdings Limited’s address is The Lennox Building, 50 Richmond Street South, Dublin D02 FK02, Ireland. |
(6) | Consists of 2,153,520 ordinary shares issuable upon conversion of (a) 424,255 ordinary shares held by Mr. Robinson, (b) 6,447 series A preferred shares held by Mr. Robinson, (c) 2 series B preferred shares held by Mr. Robinson and (d) 1,038 ordinary shares underlying options exercisable within 60 days of December 31, 2020 held by Mr. Robinson. |
(7) | Consists of (a) 858,960 ordinary shares issuable upon conversion of (i) 170,730 ordinary shares held by Blackwell A&M Trust, (ii) 1,062 series A preferred shares held by Blackwell A&M Trust, (b) 828,900 ordinary shares issuable upon conversion of (i) 164,428 ordinary shares held by Mr. Blackwell and(ii) 1,350 series A preferred shares held by Mr. Blackwell and (iii) 2 series B preferred shares held by Mr. Blackwell and (c) 31,645 ordinary shares issuable upon conversion of 6,329 ordinary shares held by the Ham Trust. |
(8) | Consists of (a) 1,539,080 ordinary shares issuable upon conversion of (i) 4,599 ordinary shares held by Baker Brothers Life Sciences L.P., (ii) 49,181 series A preferred shares held by Baker Brothers Life Sciences L.P. and (iii) 254,036 series C preferred shares held by Baker Brothers Life Sciences L.P., and (b) (a) 124,175 ordinary shares issuable upon conversion of (i) 366 ordinary shares held 677, L.P., (ii) 3,931 series A preferred shares held by 667, L.P. and (iii) 20,538 series C preferred shares held by 677, L.P. |
(9) | Consists 1,595,585 ordinary shares issuable upon conversion of 27,303 ordinary shares held by Schroders UK Public Private Trust plc and 291,814 series A preferred shares held by Schroders UK Public Private Trust plc. |
(10) | Consists of 5,760 ordinary shares issuable upon conversion of (a) 1,152 ordinary shares held by Professor Sir John Bell and (b) 5,560 ordinary shares underlying options exercisable within 60 days of December 31, 2020 held by Professor Sir John Bell. |
(11) | Consists of 5,760 ordinary shares issuable upon conversion of (a) 1,152 ordinary shares and (b) 5,560 ordinary shares underlying options exercisable within 60 days of December 31, 2020. |
• | each holder of our ordinary shares is entitled to one vote per ordinary share on all matters to be voted on by shareholders generally; |
• | the holders of the ordinary shares shall be entitled to receive notice of, attend, speak and vote at our general meetings; and |
• | the holders of our ordinary shares are entitled to receive such dividends as are recommended by our directors and declared by our shareholders. |
• | the deferred shares shall not be entitled to any dividends or to any other right of participation in the income or profits of the Company; |
• | on the return of assets on a winding-up of the Company, the deferred shares shall confer on the holders thereof an entitlement to receive out of the assets of the Company available for distribution amongst |
• | the deferred shares do not entitle the holder thereof to receive notice of, or to attend, speak or vote at any general meeting, or be part of the quorum thereof as the holders of the deferred shares; and |
• | the Company shall have irrevocable authority from each holder of deferred shares to either (i) appoint any person to execute on behalf of any holder of deferred shares a transfer of all or any of those shares and/or an agreement to transfer the same (without making any payment for them) to such person or persons as the Company may determine and to execute any other documents which such person may consider necessary or desirable to effect such transfer, in each case without obtaining the sanction of the holder(s) and without any payment being made in respect of such acquisition; and (ii) to purchase all or any of the deferred shares without obtaining the consent of the holders of those shares in consideration for an amount not exceeding £1.00 in respect of all the deferred shares then being purchased. |
• | a holder of non-voting ordinary shares shall, in relation to the non-voting ordinary shares held by him or her, have no right to receive notice of, or to attend or vote at, any general meeting of shareholders save in relation to a variation of class rights of the non-voting ordinary shares; |
• | the non-voting ordinary shares shall be re-designated as ordinary shares by our board of directors, or a duly authorised committee or representative thereof, upon receipt of a re-designation notice and otherwise subject to the terms and conditions set out therein. A holder of non-voting ordinary shares shall not be entitled to have any non-voting ordinary shares re-designated as ordinary shares where such re-designation would result in such holder thereof beneficially owning (for purposes of section 13(d) of the Exchange Act), when aggregated with “affiliates” and “group” members with whom such holder is required to aggregate beneficial ownership for purposes of section 13(d) of the Exchange Act, in excess of 9.99 per cent. of any class of securities of the Company registered under the Exchange Act (which percentage may be increased or decreased on a holder-by-holder basis subject to the provisions set out therein); and |
• | the non-voting ordinary shares shall be re-designated as ordinary shares automatically upon transfer of a non-voting ordinary share by its holder to any person that is not an “affiliate” or “group member” with whom such holder is required to aggregate beneficial ownership for purposes of section 13(d) of the Exchange Act. This automatic re-designation shall only be in respect of the non-voting ordinary shares that are subject to such transfer. |
• | the name of any person, without sufficient cause, is wrongly entered in or omitted from our register of members; or |
• | there is a default or unnecessary delay in entering on the register the fact of any person having ceased to be a member or on which we have a lien, provided that such refusal does not prevent dealings in the shares taking place on an open and proper basis. |
• | Demand Registration on Form F-1 – following this offering, each holder shall be entitled to demand registration on Form F-1, provided that these demand registration rights may only be exercised by holders who hold, in the aggregate, not less than 30% of the aggregate number of shares held, immediately prior to the completion of this offering, by all holders who are party to the agreement. These demand registration rights may not be exercised more than twice. |
• | Demand Registration on Form F-3 – each holder shall be entitled to demand registration on Form F-3, if we are eligible to register shares on Form F-3, provided that these demand registration rights may only be exercised by holders who hold, in the aggregate, not less than 20% of the aggregate number of shares held, immediately prior to the completion of this offering, by all holders who are party to the agreement. These demand registration rights may not be exercised more than twice in any calendar year. |
• | Piggyback Registration – each holder shall be entitled to piggyback registration rights, subject, in the case of an underwritten offering, to customary reductions by the underwriter. |
• | Expenses – We will pay all registration expenses relating to the exercise of the registration rights above, including the reasonable fees and expenses of one legal counsel to the participating holders up to a maximum of $50,000 in the aggregate. |
• | any resolution put to the vote of a general meeting must be decided exclusively on a poll; on a poll, every shareholder who is present in person or by proxy or corporate representative shall have one vote for each share of which they are the holder. A shareholder entitled to more than one vote need not, if they vote, use all their votes or cast all the votes in the same way; and |
• | if two or more persons are joint holders of a share, then in voting on any question the vote of the senior who tenders a vote, whether in person or by proxy, shall be accepted to the exclusion of the votes of the other joint holders. For this purpose seniority shall be determined by the order in which the names of the holders stand in the share register. |
• | it is for a share which is fully paid up; |
• | it is for a share upon which the Company has no lien; |
• | it is only for one class of share; |
• | it is in favor of a single transferee or no more than four joint transferees; |
• | it is duly stamped or is duly certificated or otherwise shown to the satisfaction of the board to be exempt from stamp duty (if this is required); and |
• | it is delivered for registration to our registered office (or such other place as the board may determine), accompanied (except in the case of a transfer by a person to whom the Company is not required by law to issue a certificate and to whom a certificate has not been issued or in the case of a renunciation) by the certificate for the shares to which it relates and such other evidence as the board may reasonably require to prove the title of the transferor (or person renouncing) and the due execution of the transfer or renunciation by him or, if the transfer or renunciation is executed by some other person on his behalf, the authority of that person to do so. |
• | the quorum for such class meeting shall be two holders in person or by proxy representing not less than one-third in nominal value of the issued shares of the class (excluding any shares held in treasury); and |
• | if at any adjourned meeting of such holders a quorum is not present at the meeting, one holder of shares of the class present in person or by proxy at an adjourned meeting constitutes a quorum. |
• | the giving of any guarantee, security or indemnity in respect of money lent or obligations incurred by him or by any other person at the request of or for the benefit of our company or any of our subsidiary undertakings; |
• | the giving of any guarantee, security or indemnity in respect of a debt or obligation of our company or any of our subsidiary undertakings for which he himself has assumed responsibility in whole or in part under a guarantee or indemnity or by the giving of security; |
• | any proposal or contract relating to an offer of securities of or by our company or any of our subsidiary undertakings in which offer he is or may be entitled to participate as a holder of securities or in the underwriting or sub-underwriting of which he is to participate; |
• | any arrangement involving any other company if the director (together with any person connected with him) has an interest of any kind in that company (including an interest by holding any position in that company or by being a member of that company), unless he is to his knowledge (either directly or indirectly) the holder of or beneficially interested in one per cent or more of any class of the equity share capital of that company (calculated exclusive of any shares of that class in that company held as treasury shares) or of the voting rights available to members of that company; |
• | any arrangement for the benefit of employees of our company or any of our subsidiary undertakings which only gives him benefits which are also generally given to employees to whom the arrangement relates; |
• | any contract relating to insurance which our company is to buy or renew for the benefit of the directors or a group of people which includes directors; and |
• | a contract relating to a pension, superannuation or similar scheme or a retirement, death, disability benefits scheme or employees’ share scheme which gives the director benefits which are also generally given to the employees to whom the scheme relates. |
• | any person, together with persons acting in concert with him, acquires, whether by a series of transactions over a period of time or not, an interest in shares which (taken together with shares in which he is already interested, and in which persons acting in concert with him are interested) carry 30% or more of the voting rights of a company; or |
• | any person who, together with persons acting in concert with him, is interested in shares which in the aggregate carry not less than 30% of the voting rights of a company but does not hold shares carrying more than 50% of such voting rights and such person, or any person acting in concert with him, acquires an interest in any other shares which increases the percentage of shares carrying voting rights in which he is interested, |
• | in respect of the default shares, the relevant shareholder shall not be entitled to vote (either in person or by representative or proxy) at any general meeting or to exercise any other right conferred by a shareholding in relation to general meetings; and |
• | where the default shares represent at least 0.25% in nominal value of the issued shares of their class, (a) any dividend or other money payable in respect of the default shares shall be retained by us without liability to pay interest and/or (b) no transfers by the relevant shareholder of any default shares may be registered (unless the shareholder himself is not in default and the shareholder provides a certificate, in a form satisfactory to the directors, to the effect that after due and careful enquiry the shareholder is satisfied that none of the shares to be transferred are default shares). |
• | if, at the time that the distribution is made, the amount of its net assets (that is, the total excess of assets over liabilities) is not less than the total of its called up share capital and undistributable reserves; and |
• | if, and to the extent that, the distribution itself, at the time that it is made, does not reduce the amount of the net assets to less than that total. |
| | | England and Wales | | | Delaware | |
Number of Directors | | | Under the Companies Act, a public limited company must have at least two directors and the number of directors may be fixed by or in the manner provided in a company’s articles of association. | | | Under Delaware law, a corporation must have at least one director and the number of directors shall be fixed by or in the manner provided in the bylaws. |
| | | | | |||
Removal of Directors | | | Under the Companies Act, shareholders may remove a director without cause by an ordinary resolution (which is passed by a simple majority of those voting in person or by proxy at a general meeting) irrespective of any provisions of any service contract the director has with the Company, provided 28 clear days’ notice of the resolution has been given to the Company and its shareholders. On receipt of notice of an intended resolution to remove a director, the Company must forthwith send a copy of the notice to the director concerned. Certain other procedural requirements under the. Companies Act must also be followed such as allowing the director to make representations against his or her removal either at the meeting or in writing. | | | Under Delaware law, any director or the entire board of directors may be removed, with or without cause, by the holders of a majority of the shares then entitled to vote at an election of directors, except (a) unless the certificate of incorporation provides otherwise, in the case of a corporation whose board of directors is classified, shareholders may effect such removal only for cause, or (b) in the case of a corporation having cumulative voting, if less than the entire board of directors is to be removed, no director may be removed without cause if the votes cast against his removal would be sufficient to elect him if then cumulatively voted at an election of the entire board of directors, or, if there are classes of directors, at an election of the class of directors of which he is a part. |
| | | | | |||
Vacancies on the Board of Directors | | | Under the laws of England and Wales, the procedure by which directors, other than a company’s initial directors, are appointed is generally set out in a company’s articles of association, provided that where two or more persons | | | Under Delaware law, vacancies and newly created directorships may be filled by a majority of the directors then in office (even though less than a quorum) or by a sole remaining director unless (a) otherwise provided in the certificate of |
| | | England and Wales | | | Delaware | |
| | | are appointed as directors of a public limited company by resolution of the shareholders, resolutions appointing each director must be voted on individually. | | | incorporation or by-laws of the corporation or (b) the certificate of incorporation directs that a particular class of stock is to elect such director, in which case a majority of the other directors elected by such class, or a sole remaining director elected by such class, will fill such vacancy. | |
| | | | | |||
Annual General Meeting | | | Under the Companies Act, a public limited company must hold an annual general meeting in each six-month period following its annual accounting reference date. | | | Under Delaware law, the annual meeting of stockholders shall be held at such place, on such date and at such time as may be designated from time to time by the board of directors or as provided in the certificate of incorporation or by the bylaws. |
| | | | | |||
General Meeting | | | Under the Companies Act, a general meeting of the shareholders of a public limited company may be called by the directors. Shareholders holding at least 5% of the paid-up capital of the Company carrying voting rights at general meetings (excluding any paid up capital held as treasury shares) can require the directors to call a general meeting and, if the directors fail to do so within a certain period, may themselves (or any of them representing more than one half of the total voting rights of all of them) convene a general meeting. | | | Under Delaware law, special meetings of the stockholders may be called by the board of directors or by such person or persons as may be authorized by the certificate of incorporation or by the bylaws. |
| | | | | |||
Notice of General Meetings | | | Subject to a company’s articles of association providing for a longer period, under the Companies Act, 21 clear days’ notice must be given for an annual general meeting and any resolutions to be proposed at the meeting. Subject to a company’s articles of association providing for a longer period, at least 14 clear days’ notice is required for any other general meeting. In addition, certain matters, such as the removal of directors or auditors, require special notice, which is 28 clear days’ notice. The shareholders of a company may in all cases consent to a shorter notice period, the proportion of shareholders’ consent required being 100% of those entitled to attend and vote in the case of an annual general meeting and, in the case of any other general meeting, a majority in number of the members having a right to attend and vote at the meeting, being a | | | Under Delaware law, unless otherwise provided in the certificate of incorporation or bylaws, written notice of any meeting of the stockholders must be given to each stockholder entitled to vote at the meeting not less than 10 nor more than 60 days before the date of the meeting and shall specify the place, date, hour, and purpose or purposes of the meeting. |
| | | England and Wales | | | Delaware | |
| | | majority who together hold not less than 95% in nominal value of the shares giving a right to attend and vote at the meeting. | | | ||
| | | | | |||
Quorum | | | Subject to the provisions of a company’s articles of association, the Companies Act provides that two shareholders present at a meeting (in person, by proxy or authorized representative under the Companies Act) shall constitute a quorum for companies with more than one member. | | | The certificate of incorporation or bylaws may specify the number of shares, the holders of which shall be present or represented by proxy at any meeting in order to constitute a quorum, but in no event shall a quorum consist of less than one third of the shares entitled to vote at the meeting. In the absence of such specification in the certificate of incorporation or bylaws, a majority of the shares entitled to vote, present in person or represented by proxy, shall constitute a quorum at a meeting of stockholders. |
| | | | | |||
Proxy | | | Under the Companies Act, at any meeting of shareholders, a shareholder may designate another person to attend, speak and vote at the meeting on their behalf by proxy. | | | Under Delaware law, at any meeting of stockholders, a stockholder may designate another person to act for such stockholder by proxy, but no such proxy shall be voted or acted upon after three years from its date, unless the proxy provides for a longer period. A director of a Delaware corporation may not issue a proxy representing the director’s voting rights as a director. |
| | | | | |||
Preemptive Rights | | | Under the Companies Act, “equity securities,” being (1) shares in the Company other than shares that, with respect to dividends and capital, carry a right to participate only up to a specified amount in a distribution, referred to as “ordinary shares,” or (2) rights to subscribe for, or to convert securities into, ordinary shares, proposed to be allotted for cash must be offered first to the existing equity shareholders in the Company in proportion to the respective nominal value of their holdings, unless an exception applies or a special resolution to the contrary has been passed by shareholders in a general meeting or the articles of association provide otherwise in each case in accordance with the provisions of the Companies Act. | | | Under Delaware law, shareholders have no preemptive rights to subscribe to additional issues of stock or to any security convertible into such stock unless, and except to the extent that, such rights are expressly provided for in the certificate of incorporation. |
| | | | | |||
Authority to Allot | | | Under the Companies Act, the directors of a company must not allot shares or grant of rights to subscribe for or to convert any security into shares unless an exception applies or an ordinary resolution to the contrary has been passed by shareholders in | | | Under Delaware law, if the corporation’s charter or certificate of incorporation so provides, the board of directors has the power to authorize the issuance of stock. It may authorize capital stock to be issued for consideration consisting of cash, any |
| | | England and Wales | | | Delaware | |
| | | a general meeting or the articles of association provide otherwise in each case in accordance with the provisions of the Companies Act. | | | tangible or intangible property or any benefit to the corporation or any combination thereof. It may determine the amount of such consideration by approving a formula. In the absence of actual fraud in the transaction, the judgment of the directors as to the value of such consideration is conclusive. | |
| | | | | |||
Liability of Directors and Officers | | | Under the Companies Act, any provision, whether contained in a company’s articles of association or any contract or otherwise, that purports to exempt a director of a company, to any extent, from any liability that would otherwise attach to him in connection with any negligence, default, breach of duty or breach of trust in relation to the Company is void. Any provision by which a company directly or indirectly provides an indemnity, to any extent, for a director of the Company or of an associated company against any liability attaching to him in connection with any negligence, default, breach of duty or breach of trust in relation to the Company of which he is a director is also void except as permitted by the Companies Act, which provides exceptions for the Company to (a) purchase and maintain insurance against such liability; (b) provide a “qualifying third party indemnity” (being an indemnity against liability incurred by the director to a person other than the Company or an associated company or criminal proceedings in which he is convicted); and (c) provide a “qualifying pension scheme indemnity” (being an indemnity against liability incurred in connection with our activities as trustee of an occupational pension plan). | | | Under Delaware law, a corporation’s certificate of incorporation may include a provision eliminating or limiting the personal liability of a director to the corporation and its stockholders for damages arising from a breach of fiduciary duty as a director. However, no provision can limit the liability of a director for: • any breach of the director’s duty of loyalty to the corporation or its stockholders; • acts or omissions not in good faith or that involve intentional misconduct or a knowing violation of law; • intentional or negligent payment of unlawful dividends or stock purchases or redemptions; or • any transaction from which the director derives an improper personal benefit. |
| | | | | |||
Voting Rights | | | For a company incorporated under the laws of England and Wales, it is usual for the articles of association to provide that, unless a poll is demanded by the shareholders of a company or is required by the chairman of the meeting or our articles of association, shareholders shall vote on all resolutions on a show of hands. Under the Companies Act, a poll may be demanded by (a) not fewer than five shareholders having the right to vote on the resolution; (b) any shareholder(s) representing not less than | | | Delaware law provides that, unless otherwise provided in the certificate of incorporation, each stockholder is entitled to one vote for each share of capital stock held by such stockholder. |
| | | England and Wales | | | Delaware | |
| | | 10% of the total voting rights of all the shareholders having the right to vote on the resolution (excluding any voting rights attaching to treasury shares); or (c) any shareholder(s) holding shares in the Company conferring a right to vote on the resolution (excluding any voting rights attaching to treasury shares) being shares on which an aggregate sum has been paid up equal to not less than 10% of the total sum paid up on all the shares conferring that right. A company’s articles of association may provide more extensive rights for shareholders to call a poll. Under the laws of England and Wales, an ordinary resolution is passed on a show of hands if it is approved by a simple majority (more than 50%) of the votes cast by shareholders present (in person or by proxy) and entitled to vote. If a poll is demanded, an ordinary resolution is passed if it is approved by holders representing a simple majority of the total voting rights of shareholders present, in person or by proxy, who, being entitled to vote, vote on the resolution. Special resolutions require the affirmative vote of not less than 75% of the votes cast by shareholders present, in person or by proxy, at the meeting. If a poll is demanded, a special resolution is passed if it is approved by holders representing not less than 75% of the total voting rights of shareholders in person or by proxy who, being entitled to vote, vote on the resolution. | | | ||
| | | | | |||
Shareholder Vote on Certain Transactions | | | The Companies Act provides for schemes of arrangement, which are arrangements or compromises between a company and any class of shareholders or creditors and used in certain types of reconstructions, amalgamations, capital reorganizations, or takeovers. These arrangements require: • the approval at a shareholders’ or creditors’ meeting convened by order of the court, of a majority in number of shareholders or creditors or a class thereof representing 75% in value of the capital held by, or debt owed to, the class of shareholders or creditors, or class thereof present and voting, either | | | Generally, under Delaware law, unless the certificate of incorporation provides for the vote of a larger portion of the stock, completion of a merger, consolidation, sale, lease or exchange of all or substantially all of a corporation’s assets or dissolution requires: • the approval of the board of directors; and • approval by the vote of the holders of a majority of the outstanding stock or, if the certificate of incorporation provides for more or less than one vote per share, a majority of the votes of the |
| | | England and Wales | | | Delaware | |
| | | in person or by proxy; and • the approval of the court. | | | outstanding stock of a corporation entitled to vote on the matter. | |
| | | | | |||
Standard of Conduct for Directors | | | Under the laws of England and Wales, a director owes various statutory and fiduciary duties to the Company, including: • to act in the way he considers, in good faith, would be most likely to promote the success of the Company for the benefit of its members as a whole, and in doing so have regard (amongst other matters) to: (i) the likely consequences of any decision in the long-term, (ii) the interests of the company’s employees, (iii) the need to foster the company’s business relationships with suppliers, customers and others, (iv) the impact of the company’s operations on the community and the environment, (v) the desirability to maintain a reputation for high standards of business conduct, and (vi) the need to act fairly as between members of the company; • to avoid a situation in which he has, or can have, a direct or indirect interest that conflicts, or possibly conflicts, with the interests of the Company; • to act in accordance with our constitution and only exercise his powers for the purposes for which they are conferred; • to exercise independent judgment; • to exercise reasonable care, skill, and diligence; • not to accept benefits from a third party conferred by reason of his being a director or doing, or not doing, anything as a director; and • a duty to declare any interest that he has, whether directly or indirectly, in a proposed or existing transaction or arrangement with the Company. | | | Delaware law does not contain specific provisions setting forth the standard of conduct of a director. The scope of the fiduciary duties of directors is generally determined by the courts of the State of Delaware. In general, directors have a duty to act without self-interest, on a well-informed basis and in a manner they reasonably believe to be in the best interest of the stockholders. Directors of a Delaware corporation owe fiduciary duties of care and loyalty to the corporation and to its shareholders. The duty of care generally requires that a director act in good faith, with the care that an ordinarily prudent person would exercise under similar circumstances. Under this duty, a director must inform himself of all material information reasonably available regarding a significant transaction. The duty of loyalty requires that a director act in a manner he reasonably believes to be in the best interests of the corporation. He must not use his corporate position for personal gain or advantage. In general, but subject to certain exceptions, actions of a director are presumed to have been made on an informed basis, in good faith and in the honest belief that the action taken was in the best interests of the corporation. However, this presumption may be rebutted by evidence of a breach of one of the fiduciary duties. Delaware courts have also imposed a heightened standard of conduct upon directors of a Delaware corporation who take any action designed to defeat a threatened change in control of the corporation. In addition, under Delaware law, when the board of directors of a Delaware corporation approves the sale or break-up of a corporation, the board of directors may, in certain circumstances, have a duty to obtain the highest value reasonably available to the shareholders. |
| | | | |
| | | England and Wales | | | Delaware | |
Shareholder Litigation | | | Under the laws of England and Wales, generally, the Company, rather than its shareholders, is the proper claimant in an action in respect of a wrong done to the Company or where there is an irregularity in the Company’s internal management. Notwithstanding this general position, the Companies Act provides that (1) a court may allow a shareholder to bring a derivative claim (that is, an action in respect of and on behalf of the Company) in respect of a cause of action arising from a director’s negligence, default, breach of duty or breach of trust and (2) a shareholder may bring a claim for a court order where our affairs have been or are being conducted in a manner that is unfairly prejudicial to some of its shareholders. | | | Under Delaware law, a stockholder may initiate a derivative action to enforce a right of a corporation if the corporation fails to enforce the right itself. The complaint must: • state that the plaintiff was a stockholder at the time of the transaction of which the plaintiff complains or that the plaintiffs shares thereafter devolved on the plaintiff by operation of law; and • allege with particularity the efforts made by the plaintiff to obtain the action the plaintiff desires from the directors and the reasons for the plaintiff’s failure to obtain the action; or • state the reasons for not making the effort. Additionally, the plaintiff must remain a stockholder through the duration of the derivative suit. The action will not be dismissed or compromised without the approval of the Delaware Court of Chancery. |
• | we do not timely request that the rights be distributed to you or we request that the rights not be distributed to you; or |
• | we fail to deliver satisfactory documents to the depositary; or |
• | it is not reasonably practicable to distribute the rights. |
• | we do not request that the property be distributed to you or if we ask that the property not be distributed to you; or |
• | we do not deliver satisfactory documents to the depositary; or |
• | the depositary determines that all or a portion of the distribution to you is not reasonably practicable. |
• | the ordinary shares are duly authorized, validly allotted and issued, fully paid, not subject to any call for the payment of further capital and legally obtained; |
• | all preemptive (and similar) rights, if any, with respect to such ordinary shares have been validly waived, disapplied or exercised; |
• | you are duly authorized to deposit the ordinary shares; |
• | the ordinary shares presented for deposit are free and clear of any lien, encumbrance, security interest, charge, mortgage or adverse claim, and are not, and the ADSs issuable upon such deposit will not be, “restricted securities” (as defined in the deposit agreement); and |
• | the ordinary shares presented for deposit have not been stripped of any rights or entitlements. |
• | ensure that the surrendered ADR is properly endorsed or otherwise in proper form for transfer; |
• | provide such proof of identity and genuineness of signatures, and of such other matters contemplated in the deposit agreement, as the depositary deems appropriate; |
• | comply with applicable laws and regulations, including regulations imposed by us and the depositary consistent with the deposit agreement, the ADR and applicable law; |
• | provide any transfer stamps required by the State of New York or the United States; and |
• | pay all applicable fees, charges, expenses, taxes and other government charges payable by ADR holders pursuant to the terms of the deposit agreement, upon the transfer of ADRs. |
• | temporary delays that may arise because (1) the transfer books for the ordinary shares or ADSs are closed, or (2) ordinary shares are immobilized on account of a shareholders’ meeting or a payment of dividends; |
• | obligations to pay fees, taxes and similar charges; or |
• | restrictions imposed because of laws or regulations applicable to ADSs or the withdrawal of securities on deposit. |
• | In the event of voting by show of hands, the depositary will vote (or cause the custodian to vote) all ordinary shares held on deposit at that time in accordance with the voting instructions received from a majority of holders of ADSs who provide timely voting instructions. |
• | In the event of voting by poll, the depositary will vote (or cause the custodian to vote) the ordinary shares held on deposit in accordance with the voting instructions received from the holders of ADSs. |
Service | | | Fee |
Issuance of ADSs (e.g., an issuance of ADS upon a deposit of ordinary shares or upon a change in the ADS(s)-to-ordinary shares ratio, or for any other reason), excluding ADS issuances as a result of distributions of ordinary shares | | | Up to $0.05 per ADS issued |
| | | ||
Cancellation of ADSs (e.g., a cancellation of ADSs for delivery of deposited property or upon a change in the ADS(s)-to-ordinary shares ratio, or for any other reason) | | | Up to $0.05 per ADS cancelled |
| | | ||
Distribution of cash dividends or other cash distributions (e.g., upon a sale of rights and other entitlements) | | | Up to $0.05 per ADS held |
| | | ||
Distribution of ADSs pursuant to (i) share dividends or other distributions, or (ii) exercise of rights to purchase additional ADSs | | | Up to $0.05 per ADS held |
| | | ||
Distribution of securities other than ADSs or rights to purchase additional ADSs (e.g., upon a spin-off) | | | Up to $0.05 per ADS held |
| | | ||
ADS services | | | Up to $0.05 per ADS held on the applicable record date(s) established by the depositary |
• | taxes (including applicable interest and penalties) and other governmental charges; |
• | the registration fees as may from time to time be in effect for the registration of ordinary shares on the share register and applicable to transfers of ordinary shares to or from the name of the custodian, the depositary or any nominees upon the making of deposits and withdrawals, respectively; |
• | certain cable, telex and facsimile transmission and delivery expenses; |
• | the expenses and charges incurred by the depositary in the conversion of foreign currency; |
• | the fees and expenses incurred by the depositary in connection with compliance with exchange control regulations and other regulatory requirements applicable to ordinary shares, ADSs and ADRs; and |
• | the fees and expenses incurred by the depositary, the custodian, or any nominee in connection with the servicing or delivery of deposited property. |
• | We and the depositary are obligated only to take the actions specifically stated in the deposit agreement without negligence or bad faith. |
• | The depositary disclaims any liability for any failure to carry out voting instructions, for any manner in which a vote is cast or for the effect of any vote, provided it acts in good faith and in accordance with the terms of the deposit agreement. |
• | The depositary disclaims any liability for any failure to accurately determine the lawfulness or practicality of any action, for the content of any document forwarded to you on our behalf or for the accuracy of any translation of such a document, for the investment risks associated with investing in ordinary shares, for the validity or worth of the ordinary shares, for any tax consequences that result from the ownership of ADSs or other deposited property, for the credit-worthiness of any third party, for allowing any rights to lapse under the terms of the deposit agreement, for the timeliness of any of our notices or for our failure to give notice or for any act or omission of or information provided by DTC or any DTC participant. |
• | The depositary shall not be liable for acts or omissions of any successor depositary in connection with any matter arising wholly after the resignation or removal of the depositary. |
• | We and the depositary will not be obligated to perform any act that is inconsistent with the terms of the deposit agreement. |
• | We and the depositary disclaim any liability if we or the depositary are prevented or forbidden from or subject to any civil or criminal penalty or restraint on account of, or delayed in, doing or performing any act or thing required by the terms of the deposit agreement, by reason of any provision, present or future of any law or regulation, including regulations of any stock exchange or by reason of present or future provisions of our articles of association, or any provision of or governing the securities on deposit, or by reason of any act of God or war or other circumstances beyond our or the depositary’s control. |
• | We and the depositary disclaim any liability by reason of any exercise of, or failure to exercise, any discretion provided for in the deposit agreement or in our articles of association or in any provisions of or governing the securities on deposit. |
• | We and the depositary further disclaim any liability for any action or inaction in reliance on the advice or information received from legal counsel, accountants, any person presenting ordinary shares for deposit, any holder of ADSs or authorized representatives thereof, or any other person believed by either of us in good faith to be competent to give such advice or information. |
• | We and the depositary also disclaim liability for the inability by any ADS holder or beneficiary owner to benefit from any distribution, offering, right or other benefit that is made available to holders of ordinary shares but is not, under the terms of the deposit agreement, made available to you. |
• | We and the depositary may rely without any liability upon any written notice, request or other document believed to be genuine and to have been signed or presented by the proper parties. |
• | We and the depositary also disclaim liability for any consequential or punitive damages for any breach of the terms of the deposit agreement. |
• | We and the depositary disclaim liability arising out of losses, liabilities, taxes, charges or expenses resulting from the manner in which a holder or beneficial owner of ADSs holds ADSs, including resulting from holding ADSs through a brokerage account. |
• | No disclaimer of any Securities Act liability is intended by any provision of the deposit agreement. |
• | Nothing in the deposit agreement gives rise to a partnership or joint venture, or establishes a fiduciary relationship, among us, the depositary and you as ADS holder. |
• | Nothing in the deposit agreement precludes Citibank (or its affiliates) from engaging in transactions in which parties adverse to us or the ADS owners have interests, and nothing in the deposit agreement obligates Citibank to disclose those transactions, or any information obtained in the course of those transactions, to us or to the ADS owners, or to account for any payment received as part of those transactions. |
• | Convert the foreign currency to the extent practical and lawful and distribute the U.S. dollars to the ADS holders for whom the conversion and distribution is lawful and practical. |
• | Distribute the foreign currency to ADS holders for whom the distribution is lawful and practical. |
• | Hold the foreign currency (without liability for interest) for the applicable ADS holders. |
• | the restricted securities have been held for at least six months, including the holding period of any prior owner other than one of our affiliates; |
• | we have been subject to the Exchange Act periodic reporting requirements for at least 90 days before the sale; and |
• | we are current in our Exchange Act reporting at the time of sale. |
• | 1% of the number of ordinary shares then outstanding, being represented by ADSs or otherwise, which will equal approximately 407,412 ordinary shares immediately after the closing of this offering based on the number of ordinary shares outstanding as of December 31, 2020; or |
• | the average weekly trading volume of our ADSs on the Nasdaq Global Market during the four calendar weeks preceding the filing of a notice on Form 144 with respect to the sale. |
• | banks, insurance companies, and certain other financial institutions; |
• | U.S. expatriates and certain former citizens or long-term residents of the United States; |
• | dealers or traders in securities who use a mark-to-market method of tax accounting; |
• | persons holding ordinary shares or ADSs as part of a hedging transaction, “straddle,” wash sale, conversion transaction or integrated transaction or persons entering into a constructive sale with respect to ordinary shares or ADSs; |
• | persons whose “functional currency” for U.S. federal income tax purposes is not the U.S. dollar; |
• | brokers, dealers or traders in securities, commodities or currencies; |
• | tax-exempt entities or government organizations; |
• | S corporations, partnerships, or other entities or arrangements classified as partnerships for U.S. federal income tax purposes (and investors therein); |
• | regulated investment companies or real estate investment trusts; |
• | persons who acquired our ordinary shares or ADSs pursuant to the exercise of any employee stock option or otherwise as compensation; |
• | persons holding shares or ADSs in connection with a trade or business outside the United States; |
• | persons that own or are deemed to own ten percent or more of our shares (by vote or value); and |
• | persons holding our ordinary shares or ADSs in connection with a trade or business, permanent establishment, or fixed base outside the United States. |
(1) | an individual who is a citizen or resident of the United States; |
(2) | a corporation, or other entity taxable as a corporation for U.S. federal income tax purposes, created or organized in or under the laws of the United States, any state therein or the District of Columbia; |
(3) | an estate the income of which is subject to U.S. federal income taxation regardless of its source; or |
(4) | a trust if (1) a U.S. court is able to exercise primary supervision over the administration of the trust and one or more U.S. persons have authority to control all substantial decisions of the trust or (2) the trust has a valid election to be treated as a U.S. person under applicable U.S. Treasury Regulations. |
• | the excess distribution or gain will be allocated ratably over a U.S. Holder’s holding period for the ordinary shares or ADSs; |
• | the amount allocated to the taxable year of the disposition or distribution (as applicable), and any taxable year prior to the first taxable year in which we became a PFIC, will be treated as ordinary income; and |
• | the amount allocated to each other year will be subject to the highest tax rate in effect for that year and the interest charge generally applicable to underpayments of tax will be imposed on the resulting tax attributable to each such year. |
• | persons who are connected with the company; |
• | financial institutions; |
• | insurance companies; |
• | charities or tax-exempt organizations; |
• | collective investment schemes; |
• | pension schemes; |
• | market makers, intermediaries, brokers or dealers in securities; |
• | persons who have (or are deemed to have) acquired their ADSs by virtue of an office or employment or who are or have been officers or employees of the company or any of its affiliates; and |
• | individuals who are subject to U.K. taxation on a remittance basis. |
Underwriters | | | Number of ADSs |
Goldman Sachs & Co. LLC | | | |
J.P. Morgan Securities LLC | | | |
Jefferies LLC | | | |
Total | | | 8,333,333 |
| | | No Exercise | | | Full Exercise | |
Per ADS | | | $ | | | $ |
Total | | | $ | | | $ |
• | the ADSs offered hereby; |
• | as a result of the exercise of share options or the award of share options or restricted shares existing on, or upon the conversion or exchange of convertible or exchangeable securities outstanding as of the date of the underwriting agreement; |
• | pursuant to the corporate reorganization; |
• | the grant of awards pursuant to certain employee equity-based compensation plans; |
• | the filing of a registration statement on Form S-8 in connection with the registration of ordinary shares issuable under any employee equity based compensation plan; and |
• | the issuance of up to 5% of the outstanding ordinary shares in connection with the acquisition of the assets of, or a majority or controlling portion of the equity of, or a joint venture, provided however, that each recipient of any ordinary shares pursuant to this exception shall have executed a lock-up agreement prior to such issuance or sale, as the case may be. |
• | as a bona fide gift or gifts or charitable contribution; |
• | to any trust for the direct or indirect benefit of the lock-up party or the immediate family of the lock-up party; |
• | with the prior written consent of the representatives on behalf of the underwriters; |
• | by will or intestacy; |
• | to any corporation, partnership limited liability company or other business entity, all of the beneficial ownership interests of which, in each such case, are held by the lock-up party or any member of the lock-up party’s immediate family; |
• | by operation of law, including pursuant to a domestic order or negotiated divorce settlement; |
• | (i) the exercise of options or other similar awards or the vesting or settlement of awards granted pursuant to the our equity incentive plans as described herein (including the delivery and receipt of ordinary shares or ADSs, other awards or any securities convertible into or exercisable or exchangeable for ordinary shares or ADSs in connection with such exercise, vesting or settlement), or (ii) the transfer or disposition of ordinary shares or ADSs or any securities convertible into ordinary shares or ADSs by the lock-up party to us (or the purchase and cancellation of same by us) upon a vesting or settlement event of the our securities or upon the exercise of options to purchase the our securities on a “cashless” or “net exercise” basis to the extent permitted by the instruments representing such options pursuant to our share option plan, equity incentive plan, share purchase plan or other equity incentive arrangement as described herein; |
• | to us to the extent required to realize sufficient funds to satisfy the exercise price and/or any income, employment tax and/or social security withholding and remittance obligations upon the vesting or exercise of an option or other award granted under a share option plan, equity incentive plan, share purchase plan or other equity incentive arrangement by us described herein or the conversion or exercise of a warrant described herein; |
• | to us pursuant to any contractual arrangement in effect on the date the lock-up party entered into the lock-up agreement and described herein that provides for the repurchase of the lock-up party’s ordinary shares or ADSs by the us in connection with the termination of the lock-up party’s employment or other service relationship with us or the lock-up party’s failure to meet certain conditions set out upon receipt of such ordinary shares or ADSs; |
• | in connection with the corporate reorganization as described herein and consummated before, or at the same time as, the closing of this offering; |
• | acquired in the offering, or in open market transactions following the offering; |
• | as part of a distribution, transfer or disposition without consideration by the lock-up party to its limited or general partners, members, stockholders or affiliates (as defined under Rule 12b-2 of the Exchange Act); |
• | in connection with the establishment or amendment of a trading plan pursuant to Rule 10b5-1 under the Exchange Act, provided that (A) the lock-up party does not otherwise voluntarily effect any public filing or report regarding the establishment of such plan during the lock-up period and (B) no sale or other transfer of ordinary shares or ADSs pursuant to such plan may occur during the lock-up period; |
• | pursuant to a bona fide third-party tender offer, merger, takeover offer consolidation, scheme of arrangement or other similar transaction approved by the our board of directors and made with or offered to all holders of the our ordinary shares and ADSs resulting in a change in the ownership of 90% of our voting capital stock that is made or offered after the offering, provided that, in the event that such change of control is not completed, the lock-up party’s ordinary shares and ADSs shall remain subject to the restrictions contained in the lock-up agreement and title to the lock-up party’s ordinary shares and ADSs shall remain with the lock-up party; |
• | through the deposit of ordinary shares with our ADS depositary in exchange for the issuance of ADSs, or the cancellation of ADSs and withdrawal of underlying ordinary shares; and |
• | pursuant to an existing agreement by one of our beneficial owners to sell up to 15,194,100 ordinary shares (or such equivalent number of ordinary shares thereof following the corporate reorganization); provided, however, that such transferee shall agree to be bound in writing by the same restrictions set forth herein; provided, further, that such beneficial owner and such transferee shall also agree to the publicity limitations relating to such transfer, as further described in the lock-up agreement. |
(a) | to any legal entity which is a qualified investor as defined under Article 2 of the Prospectus Regulation; |
(b) | to fewer than 150 natural or legal persons (other than qualified investors as defined under Article 2 of the Prospectus Regulation), subject to obtaining the prior consent of the Representatives for any such offer; or |
(c) | in any other circumstances falling within Article 1(4) of the Prospectus Regulation, |
(a) | to any legal entity which is a qualified investor as defined under Article 2 of the U.K. Prospectus Regulation; |
(b) | to fewer than 150 natural or legal persons (other than qualified investors as defined under Article 2 of the U.K. Prospectus Regulation), subject to obtaining the prior consent of the Representatives for any such offer; or |
(c) | in any other circumstances falling within Section 86 of the FSMA. |
Expense | | | Amount |
SEC registration fee | | | $26,139 |
Nasdaq initial listing fee | | | $150,000 |
FINRA filing fee | | | $36,437 |
Printing expenses | | | $226,500 |
Legal fees and expenses | | | $2,500,000 |
Accounting fees and expenses | | | $1,320,387 |
Miscellaneous fees and expenses | | | $140,537 |
Total | | | $4,400,000 |
• | recognize or enforce judgments of U.S. courts obtained against us or our directors or officers predicated upon the civil liabilities provisions of the securities laws of the United States or any state in the United States; or |
• | entertain original actions brought in England and Wales against us or our directors or officers predicated upon the securities laws of the United States or any state in the United States. |
• | the relevant U.S. court had jurisdiction over the original proceedings according to English conflicts of laws principles at the time when proceedings were initiated; |
• | England and Wales courts had jurisdiction over the matter on enforcement and we either submitted to such jurisdiction or were resident or carrying on business within such jurisdiction and were duly served with process; |
• | the U.S. judgment was final and conclusive on the merits in the sense of being final and unalterable in the court that pronounced it and being for a definite sum of money; |
• | the judgment given by the courts was not in respect of penalties, taxes, fines or similar fiscal or revenue obligations (or otherwise based on a U.S. law that an English court considers to relate to a penal, revenue or other public law); |
• | the judgment was not procured by fraud; |
• | the judgment was not obtained following a breach of a jurisdictional or arbitrational clause, unless with the agreement of the defendant as the defendant’s subsequent submission to the jurisdiction of the court; |
• | recognition or enforcement of the judgment in England and Wales would not be contrary to public policy or the Human Rights Act 1998; |
• | the proceedings pursuant to which judgment was obtained were not contrary to natural justice; |
• | the U.S. judgment was not arrived at by doubling, trebling or otherwise multiplying a sum assessed as compensation for the loss or damages sustained and not being otherwise in breach of Section 5 of the U.K. Protection of Trading Interests Act 1980, or is a judgment based on measures designated by the Secretary of State under Section 1 of that Act; |
• | there is not a prior decision of an English court or the court of another jurisdiction on the issues in question between the same parties; and |
• | the English enforcement proceedings were commenced within the limitation period. |
| | | page | |
Audited Financial Statement of Immunocore Holdings Limited | | | |
| | | ||
| | | ||
| | | ||
Audited Consolidated Financial Statements of Immunocore Limited | | | |
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
Unaudited Condensed Consolidated Interim Financial Statements of Immunocore Limited | | | |
| | | ||
| | | ||
| | | ||
| | | ||
| | |
| | | January 7, 2021 | |
| | | £ | |
Total assets | | | — |
Equity | | | |
Share capital £0.0001 par value, one share authorized, called up and fully paid | | | — |
Share premium | | | — |
Total equity | | | — |
Total liabilities | | | — |
Total equity and liabilities | | | — |
| | | Notes | | | 2019 £’000 | | | 2018 £’000 | |
Revenue | | | 3 | | | 25,669 | | | 23,654 |
Total revenue | | | | | 25,669 | | | 23,654 | |
| | | | | | | ||||
Other operating income | | | 6 | | | 185 | | | 622 |
Research and development costs | | | 4 | | | (99,991) | | | (83,575) |
Administrative expenses | | | 4 | | | (44,183) | | | (34,156) |
Operating loss | | | | | (118,320) | | | (93,455) | |
| | | | | | | ||||
Other income | | | | | — | | | 4,979 | |
Finance income | | | 7 | | | 1,510 | | | 1,140 |
Finance costs | | | 8 | | | (9,379) | | | (842) |
Non-operating (expense) / income | | | | | (7,869) | | | 5,277 | |
| | | | | | | ||||
Loss before taxation | | | | | (126,189) | | | (88,178) | |
Income tax credit | | | 9 | | | 22,258 | | | 16,548 |
Loss for the year | | | | | (103,931) | | | (71,630) | |
| | | | | | | ||||
Other comprehensive (expense) / income | | | | | | | |||
Other comprehensive (expense) / income that are or may be reclassified to profit or loss in subsequent periods (net of tax): | | | | | | | |||
Exchange differences on translation of foreign operations | | | | | (99) | | | 72 | |
Income tax effect relating to the components of other comprehensive income | | | 9 | | | — | | | 3,634 |
Total other comprehensive (expense) / income for the year, net of tax | | | | | (99) | | | 3,706 | |
| | | | | | | ||||
Total comprehensive loss for the year, net of tax | | | | | (104,030) | | | (67,924) | |
Basic and diluted loss per share | | | 10 | | | (0.02) | | | (0.02) |
| | | Notes | | | 2019 £’000 | | | 2018 £’000 | |
Non-current assets | | | | | | | |||
Intangible assets | | | 11 | | | — | | | 318 |
Property, plant and equipment | | | 12 | | | 54,880 | | | 20,874 |
Investment in sub-lease | | | 13 | | | 591 | | | — |
Other non-current financial assets | | | 14 | | | 4,390 | | | 2,532 |
Deferred tax asset | | | 9 | | | 1,507 | | | 872 |
Total non-current assets | | | | | 61,368 | | | 24,596 | |
Current assets | | | | | | | |||
Trade and other receivables | | | 16 | | | 9,639 | | | 13,738 |
Tax receivable | | | | | 40,410 | | | 32,339 | |
Embedded derivative assets | | | 23 | | | 266 | | | 719 |
Cash and cash equivalents | | | 17 | | | 73,966 | | | 124,385 |
Total current assets | | | | | 124,281 | | | 171,181 | |
Total assets | | | | | 185,649 | | | 195,777 | |
Equity | | | | | | | |||
Share capital | | | 18 | | | — | | | — |
Share premium | | | 18 | | | 283,250 | | | 224,087 |
Foreign currency translation reserve | | | 18 | | | (32) | | | 67 |
Share-based payment reserve | | | 18, 22 | | | 10,659 | | | 7,603 |
Accumulated deficit | | | | | (279,106) | | | (175,175) | |
Total equity | | | | | 14,771 | | | 56,582 | |
| | | | | | | ||||
Non-current liabilities | | | | | | | |||
Interest-bearing loans and borrowings | | | 19 | | | — | | | 18,878 |
Deferred liabilities | | | 19 | | | 47,961 | | | 70,665 |
Lease liabilities | | | 13 | | | 38,299 | | | — |
Provisions | | | 20 | | | 105 | | | 45 |
Total non-current liabilities | | | | | 86,365 | | | 89,588 | |
| | | | | | | ||||
Current liabilities | | | | | | | |||
Interest-bearing loans and borrowings | | | 21 | | | 19,157 | | | — |
Trade and other payables | | | 21 | | | 29,501 | | | 19,555 |
Deferred liabilities | | | 21 | | | 28,522 | | | 29,741 |
Tax payable | | | 21 | | | 72 | | | 139 |
Lease liabilities | | | 13 | | | 1,951 | | | — |
Derivative liabilities | | | 23 | | | 5,127 | | | — |
Provisions | | | 20 | | | 183 | | | 172 |
Total current liabilities | | | | | 84,513 | | | 49,607 | |
Total liabilities | | | | | 170,878 | | | 139,195 | |
Total equity and liabilities | | | | | 185,649 | | | 195,777 |
| | | Notes | | | Share capital £’000 | | | Share premium £’000 | | | Foreign currency translation reserve £’000 | | | Available- for-sale reserve £’000 | | | Share- based payment reserve £’000 | | | Accumulated deficit £’000 | | | Total equity £’000 | |
At January 1, 2018 | | | | | — | | | 223,986 | | | (5) | | | 14,962 | | | 6,812 | | | (122,016) | | | 123,739 | |
Loss for the year | | | | | — | | | — | | | — | | | — | | | — | | | (71,630) | | | (71,630) | |
Reclassification on sale of asset held for sale | | | 15 | | | — | | | — | | | — | | | (18,471) | | | — | | | 18,471 | | | — |
Other comprehensive income | | | | | — | | | — | | | 72 | | | 3,509 | | | 125 | | | — | | | 3,706 | |
Total comprehensive loss for the year | | | | | — | | | — | | | 72 | | | (14,962) | | | 125 | | | (53,159) | | | (67,924) | |
Issue of share capital | | | 18 | | | — | | | 101 | | | — | | | — | | | — | | | — | | | 101 |
Equity-settled share-based payment transactions | | | 18, 22 | | | — | | | — | | | — | | | — | | | 666 | | | — | | | 666 |
At December 31, 2018 | | | | | — | | | 224,087 | | | 67 | | | — | | | 7,603 | | | (175,175) | | | 56,582 | |
Loss for the year | | | | | — | | | — | | | — | | | — | | | — | | | (103,931) | | | (103,931) | |
Other comprehensive loss | | | | | — | | | — | | | (99) | | | — | | | — | | | — | | | (99) | |
Total comprehensive loss for the year | | | | | — | | | — | | | (99) | | | — | | | — | | | (103,931) | | | (104,030) | |
Issue of share capital | | | 18 | | | — | | | 59,163 | | | — | | | — | | | — | | | — | | | 59,163 |
Equity-settled share-based payment transactions | | | 18, 22 | | | — | | | — | | | — | | | — | | | 3,056 | | | — | | | 3,056 |
At December 31, 2019 | | | | | — | | | 283,250 | | | (32) | | | — | | | 10,659 | | | (279,106) | | | 14,771 |
| | | Notes | | | 2019 £’000 | | | 2018 £’000 | |
Cash flows from operating activities | | | | | | | |||
Loss for the year | | | | | (103,931) | | | (71,630) | |
Adjustments for: | | | | | | | |||
Depreciation of property, plant and equipment | | | 12 | | | 9,003 | | | 6,410 |
Amortization of intangible assets | | | 11 | | | 210 | | | 297 |
Write-off of intangible assets | | | | | 306 | | | 170 | |
Loss on disposal of property, plant and equipment | | | 4 | | | 3 | | | 135 |
Gross gain from sale of equity investment | | | | | — | | | (5,204) | |
Net finance costs / (income) | | | | | 7,867 | | | (298) | |
Movement in provisions and other charges | | | 20 | | | 71 | | | (50) |
Foreign exchange translation differences | | | | | (618) | | | 1,157 | |
Equity settled share-based payment expenses | | | 22 | | | 3,056 | | | 666 |
Taxation charge | | | 9 | | | (22,258) | | | (16,548) |
Working capital adjustments: | | | | | | | |||
Decrease/(increase) in trade and other receivables | | | 16 | | | 1,828 | | | (1,522) |
Increase in trade and other payables | | | 21 | | | 9,946 | | | 5,300 |
(Decrease)/increase in deferred liabilities | | | 19, 21 | | | (21,866) | | | 63,797 |
Cash used in operations | | | | | (116,383) | | | (17,320) | |
Bank interest received on cash and cash equivalents | | | 7 | | | 1,525 | | | 760 |
Net taxation received | | | 9 | | | 13,482 | | | (66) |
Net cash used in operating activities | | | | | (101,376) | | | (16,626) | |
Cash flows from investing activities | | | | | | | |||
Proceeds from sale of property, plant and equipment | | | 12 | | | 82 | | | — |
Gross proceeds from disposal of equity investment | | | 15 | | | — | | | 27,451 |
Purchase of property, plant and equipment | | | 12 | | | (4,078) | | | (3,486) |
Purchase of intangible assets | | | 11 | | | (198) | | | (51) |
Proceeds from sub-leases | | | | | 57 | | | — | |
Investment in short and long-term treasury deposits | | | | | — | | | 34,100 | |
Net cash flows used in investing activities | | | | | (4,137) | | | 58,014 | |
Cash flows from financing activities | | | | | | | |||
Proceeds from exercise of share options | | | 22 | | | 27 | | | 101 |
Gross proceeds from issue of share capital | | | 18 | | | 59,874 | | | — |
Costs from issue of share capital | | | | | (738) | | | ||
Repayment of lease liabilities | | | 13 | | | (4,036) | | | — |
Net cash flows from financing activities | | | | | 55,127 | | | 101 | |
Increase/(decrease) in net cash and cash equivalents | | | | | (50,386) | | | 41,489 | |
Net foreign exchange difference on cash held | | | | | (33) | | | 13 | |
Cash and cash equivalents at beginning of the year | | | | | 124,385 | | | 82,883 | |
Cash and cash equivalents at end of the year | | | | | 73,966 | | | 124,385 |
• | the Group has key worker status which allows continuity of providing services throughout a prolonged lockdown period; |
• | the Group has a track record of meeting expectations under its collaboration agreements and meeting expected milestones within the contracted timeframe; |
• | the Group’s history of being able to access equity and loan financing as and when needed; and |
• | the Group’s ability and history to control capital expenditure costs and lower other operational spend, as necessary. |
• | whether achievement of a development milestone is highly susceptible to factors outside the entity’s influence, such as milestones involving the judgment or actions of third parties, including regulatory bodies or the customer; |
• | whether the uncertainty about the achievement of the milestone is not expected to be resolved for a long period of time; |
• | whether the Company can reasonably predict that a milestone will be achieved based on previous experience; and. |
• | the complexity and inherent uncertainty underlying the achievement of the milestone. |
• | adjustments arising from a change in the estimate of when the performance obligation will have been completed; |
• | adjustment to revenue that affects deferred revenue; |
• | a change in the estimate of the transaction price due to changes in the assessment of whether variable consideration is constrained because it is not considered probable of being received; and |
• | the recognition of revenue. |
• | the data generated from the Group’s research and development programs; |
• | the future operating performance, prospects and business strategy; |
• | the material risks related to the Group’s business and industry; |
• | the lack of an active public market for the Group’s ordinary and convertible preferred shares; |
• | the market performance of publicly traded companies in the life science and biotechnology sectors; |
• | the prices at which the Group issued ordinary and preferred shares and the superior rights and preferences of the preferred shares relative to the ordinary shares at the time of each grant; and |
• | the likelihood of achieving a liquidity events for the holders of our ordinary shares, series A and B shares and Growth Shares, such as an IPO, given prevailing market conditions. |
• | Level 1: quoted prices (unadjusted) in active markets for identical assets or liabilities. |
• | Level 2: inputs other than quoted prices included in Level 1 that are observable for the asset or liability, either directly (i.e. as prices) or indirectly (i.e. derived from prices). |
• | Level 3: inputs for the asset or liability that are not based on observable market data (unobservable inputs). |
| | | December 31, 2018 as previously reported £’000 | | | IFRS 16 adjustments £’000 | | | January 1, 2019 as adjusted £’000 | |
Non-current assets | | | 24,596 | | | 44,984 | | | 69,580 |
Current assets | | | 171,181 | | | (486) | | | 170,695 |
Current liabilities | | | (49,607) | | | (828) | | | (50,435) |
Non-current liabilities | | | (89,588) | | | (43,670) | | | (133,258) |
Total net assets | | | 56,582 | | | — | | | 56,582 |
• | Applied a single discount rate to a portfolio of leases with similar characteristics. |
• | Applied the exemption not to recognize right of use assets and liabilities for assets with less than 12 months of lease term. |
• | Excluded initial direct costs from measuring the right of use asset at the date of initial application. |
| | | 2019 £’000 | | | 2018 £’000 | |
GlaxoSmithKline | | | 5,753 | | | 6,079 |
Eli Lilly | | | 819 | | | 8,561 |
Genentech | | | 19,097 | | | 1,461 |
MedImmune | | | — | | | 7,553 |
| | | 25,669 | | | 23,654 |
United Kingdom | | | 5,753 | | | 6,079 |
United States | | | 19,916 | | | 17,575 |
| | | 25,669 | | | 23,654 |
| | | 2019 £’000 | | | 2018 £’000 | |
Current deferred income (see Note 21) | | | 28,457 | | | 29,437 |
Non-current deferred income (see Note 19) | | | 47,961 | | | 68,795 |
| | | 76,418 | | | 98,232 |
| | | 2019 £’000 | | | 2018 £’000 | |
Research and development costs | | | 99,991 | | | 83,575 |
Loss on disposal of property, plant and equipment | | | 3 | | | 135 |
Loss on write-offs of intangible fixed assets | | | 306 | | | 170 |
Depreciation of property, plant and equipment (see Note 12) | | | 9,003 | | | 6,410 |
Amortization of intangible assets (see Note 11) | | | 210 | | | 297 |
Operating lease expense (see Note 13) | | | 486 | | | 4,205 |
Operating lease income (see Note 6) | | | 185 | | | (622) |
Realized foreign exchange (gains)/loss | | | 189 | | | (1,341) |
| | | 2019 No. of employees | | | 2018 No. of employees | |
Research | | | 284 | | | 299 |
Development | | | 108 | | | 95 |
Corporate | | | 67 | | | 67 |
Total | | | 459 | | | 461 |
| | | 2019 £’000 | | | 2018 £’000 | |
Wages and salaries | | | 31,920 | | | 29,501 |
Social security costs | | | 2,767 | | | 2,731 |
Share-based payments (see Note 22) | | | 3,056 | | | 666 |
Contributions to defined contribution plans (see Note 24) | | | 1,213 | | | 981 |
| | | 38,956 | | | 33,879 |
| | | 2019 £’000 | | | 2018 £’000 | |
Rental income | | | 185 | | | 622 |
| | | 185 | | | 622 |
| | | 2019 £’000 | | | 2018 £’000 | |
Bank interest on cash and cash equivalents | | | 1,386 | | | 550 |
Interest on short-term deposits | | | — | | | 272 |
Gain on entering into sub-leases on leasehold properties | | | 115 | | | — |
Lease interest income | | | 9 | | | — |
Gain from change in fair value of embedded derivative asset | | | — | | | 318 |
| | | 1,510 | | | 1,140 |
| | | 2019 £’000 | | | 2018 £’000 | |
Interest on lease liabilities | | | 2,947 | | | — |
Interest expenses on financial liabilities measured at amortized cost | | | 849 | | | 842 |
Loss from change in fair value of embedded derivative asset | | | 454 | | | — |
Loss from change in fair value of derivative liability | | | 5,127 | | | — |
Other finance costs | | | 2 | | | — |
| | | 9,379 | | | 842 |
| | | 2019 £’000 | | | 2018 £’000 | |
Profit or loss | | | | | ||
Current tax: | | | | | ||
R&D tax credit for the year | | | (21,767) | | | (18,486) |
Tax related to share-based compensation plans | | | — | | | 125 |
Foreign corporation tax on profits for the year | | | 152 | | | 139 |
Adjustments in respect of prior years | | | 43 | | | — |
Total current tax | | | (21,572) | | | (18,222) |
Deferred tax: | | | | | ||
Originating and reversal of timing differences, including adjustments in respect of prior years | | | (686) | | | 1,674 |
Total deferred tax | | | (686) | | | 1,674 |
Total income tax credit | | | (22,258) | | | (16,548) |
| | | 2019 £’000 | | | 2018 £’000 | |
Tax related to items recognized in other comprehensive income during the year: | | | | | ||
Current tax related to share-based compensation plans | | | — | | | (125) |
Deferred tax on fair value movements of available-for-sale financial assets | | | — | | | (3,509) |
Tax charged to other comprehensive income | | | — | | | (3,634) |
| | | 2019 £’000 | | | 2018 £’000 | |
Loss before tax | | | (126,189) | | | (88,178) |
Tax credit using the UK Corporation tax rate of 19% (2018: 19%) | | | (23,976) | | | (16,754) |
| | | | | |||
Effect of: | | | | | ||
Non-deductible expenses | | | 13,148 | | | 629 |
Income not taxable for tax purposes | | | — | | | (954) |
Chargeable gain on sale of assets held for sale | | | — | | | 4,359 |
Other permanent differences | | | (1) | | | (38) |
Additional deduction for R&D expenditure | | | (29,365) | | | (13,691) |
Surrender of tax losses for R&D tax credit refund | | | 28,523 | | | 24,223 |
R&D expenditure credits | | | (22,602) | | | (19,215) |
Credit to other comprehensive income for share-based compensation plans | | | — | | | 125 |
Movement in deferred tax not recognized | | | 12,413 | | | 4,746 |
Adjustments to tax charge in respect of previous periods - deferred tax | | | (500) | | | — |
Adjustments to tax charge in respect of previous periods | | | 43 | | | — |
Effects of tax rates in foreign jurisdictions | | | 59 | | | 22 |
Total tax credit included in loss for the year | | | (22,258) | | | (16,548) |
| | | 2019 £’000 | | | 2018 £’000 | |
Current tax: | | | | | ||
United States: | | | | | ||
Federal | | | 100 | | | 137 |
State | | | 15 | | | 2 |
United Kingdom | | | (21,687) | | | (18,361) |
Total current tax | | | (21,572) | | | (18,222) |
Deferred tax: | | | | | ||
United States: | | | | | ||
Federal | | | (644) | | | (516) |
State | | | (42) | | | (1) |
United Kingdom | | | — | | | 2,191 |
Total deferred tax | | | (686) | | | 1,674 |
Total income tax credit | | | (22,258) | | | (16,548) |
| | | 2019 £’000 | | | 2018 £’000 | |
United States | | | — | | | — |
United Kingdom – current tax | | | — | | | (125) |
United Kingdom – deferred tax | | | — | | | (3,509) |
Tax charged to other comprehensive income | | | — | | | (3,634) |
| | | 2019 £’000 | | | 2018 £’000 | |
Loss for the year | | | (103,931) | | | (71,630) |
Basic and diluted weighted average number of shares | | | 4,459,587 | | | 4,311,778 |
Basic and diluted loss per share | | | (0.02) | | | (0.02) |
| | | Patent and trademarks £’000 | | | Computer software £’000 | | | Assets under construction £’000 | | | Total £’000 | |
Cost: | | | | | | | | | ||||
At January 1, 2018 | | | 516 | | | 828 | | | 170 | | | 1,514 |
Additions | | | — | | | 38 | | | 13 | | | 51 |
Write-offs | | | — | | | — | | | (170) | | | (170) |
Effect of foreign currency translation | | | — | | | 1 | | | — | | | 1 |
At December 31, 2018 | | | 516 | | | 867 | | | 13 | | | 1,396 |
| | | | | | | | |
| | | Patent and trademarks £’000 | | | Computer software £’000 | | | Assets under construction £’000 | | | Total £’000 | |
Additions | | | — | | | 76 | | | 122 | | | 198 |
Transferred | | | — | | | 24 | | | (24) | | | — |
Write-offs | | | — | | | (967) | | | (111) | | | (1,078) |
At December 31, 2019 | | | 516 | | | — | | | — | | | 516 |
| | | | | | | | | |||||
Amortization and impairment: | | | | | | | | | ||||
At January 1, 2018 | | | 477 | | | 304 | | | — | | | 781 |
Amortization for the year | | | 39 | | | 258 | | | — | | | 297 |
At December 31, 2018 | | | 516 | | | 562 | | | — | | | 1,078 |
| | | | | | | | | |||||
Write-offs | | | — | | | (772) | | | — | | | (772) |
Amortization for the year | | | — | | | 210 | | | — | | | 210 |
At December 31, 2019 | | | 516 | | | — | | | — | | | 516 |
| | | | | | | | | |||||
Carrying value: | | | | | | | | | ||||
At December 31, 2019 | | | — | | | — | | | — | | | — |
At December 31, 2018 | | | — | | | 305 | | | 13 | | | 318 |
At January 1, 2018 | | | 39 | | | 524 | | | 170 | | | 733 |
| | | Leasehold properties and improvements including right of use assets £’000 | | | Plant and equipment £’000 | | | Assets under construction £’000 | | | Total £’000 | |
Cost: | | | | | | | | | ||||
At January 1, 2018 | | | 7,650 | | | 22,943 | | | 3,934 | | | 34,527 |
Additions | | | 146 | | | 1,571 | | | 1,769 | | | 3,486 |
Transfers | | | 3,558 | | | 1,156 | | | (4,714) | | | — |
Effect of foreign currency translation | | | 10 | | | 7 | | | — | | | 17 |
Disposals | | | (227) | | | (38) | | | — | | | (265) |
At December 31, 2018 | | | 11,137 | | | 25,639 | | | 989 | | | 37,765 |
| | | | | | | | | |||||
Effect of adopting new accounting standards | | | 44,984 | | | — | | | — | | | 44,984 |
Additions | | | 1,112 | | | 1,150 | | | 2,713 | | | 4,975 |
Transfers | | | 1,090 | | | 41 | | | (1,131) | | | — |
Effect of foreign currency translation | | | (17) | | | (4) | | | — | | | (21) |
Remeasurements | | | (6,849) | | | — | | | — | | | (6,849) |
Disposals | | | (185) | | | (500) | | | — | | | (685) |
At December 31, 2019 | | | 51,272 | | | 26,326 | | | 2,571 | | | 80,169 |
| | | | | | | | | |||||
Depreciation and impairment: | | | | | | | | | ||||
At January 1, 2018 | | | 1,821 | | | 8,787 | | | — | | | 10,608 |
Depreciation charge for the year | | | 2,023 | | | 4,387 | | | — | | | 6,410 |
Effect of foreign currency translation | | | — | | | 3 | | | — | | | 3 |
Disposals | | | (92) | | | (38) | | | — | | | (130) |
At December 31, 2018 | | | 3,752 | | | 13,139 | | | — | | | 16,891 |
Change in accounting policies | | | — | | | — | | | — | | | — |
Depreciation charge for the year | | | 4,501 | | | 4,502 | | | — | | | 9,003 |
Effect of foreign currency translation | | | (2) | | | (3) | | | — | | | (5) |
Disposals | | | (155) | | | (445) | | | — | | | (600) |
At December 31, 2019 | | | 8,096 | | | 17,193 | | | — | | | 25,289 |
| | | | | | | | | |||||
Carrying value: | | | | | | | | | ||||
At December 31, 2019 | | | 43,176 | | | 9,133 | | | 2,571 | | | 54,880 |
At December 31, 2018 | | | 7,385 | | | 12,500 | | | 989 | | | 20,874 |
At January 1, 2018 | | | 5,829 | | | 14,156 | | | 3,934 | | | 23,919 |
• | Options to terminate the lease early at the right of the tenant |
• | Variable lease payments with a guaranteed minimum increase and capped maximum increase |
| | | 2019 £’000 | |
Balance at January 1, 2019 | | | — |
Effect of adopting new accounting standards | | | 44,984 |
Additions | | | 897 |
Remeasurements | | | (6,849) |
Depreciation charge for the year | | | (2,454) |
| | | 36,578 |
| | | 2019 £’000 | |
Less than one year | | | 4,469 |
One to five years | | | 16,834 |
More than five years | | | 45,288 |
Total undiscounted lease liabilities at December 31, 2019 | | | 66,591 |
| | | 2019 £’000 | |
Current | | | 1,951 |
Non-current | | | 38,299 |
Total lease liabilities at December 31, 2019 | | | 40,250 |
Amounts recognized in the Consolidated Statement of Loss | | | 2019 £’000 |
Interest on lease liabilities | | | 2,947 |
Expenses relating to short-term leases | | | 486 |
Expenses relating to leases of low-value assets | | | 33 |
Income from sub-leasing right-of-use-asset | | | (9) |
| | | 2019 £’000 | | | 2018 £000’s | |
Within one year | | | 73 | | | 4,329 |
After one year but not more than five years | | | — | | | 16,566 |
More than five years | | | — | | | 60,691 |
| | | 73 | | | 81,586 |
Amounts recognized in the Consolidated Statement of Cash Flows | | | 2019 £’000 |
Total cash outflow for leases | | | 4,036 |
Lease income | | | 2019 £’000 |
Operating lease income | | | 185 |
Finance lease income on the net investment in the lease | | | 9 |
Maturity analysis – undiscounted finance lease income | | | 2019 £’000 |
Less than one year | | | 317 |
One to two years | | | 317 |
Two to three years | | | 12 |
Three to four years | | | — |
Four to five years | | | — |
More than five years | | | — |
Total undiscounted finance lease income | | | 646 |
Unearned finance income | | | (39) |
Net investment in the lease | | | 607 |
Maturity analysis – undiscounted operating lease income | | | 2019 £’000 | | | 2018 £’000 |
Less than one year | | | 96 | | | 176 |
One to two years | | | 50 | | | 11 |
Two to three years | | | 12 | | | 11 |
Three to four years | | | — | | | 11 |
Four to five years | | | — | | | 11 |
More than five years | | | — | | | — |
Total undiscounted operating lease income | | | 158 | | | 220 |
Security deposits | | | 2019 £’000 | | | 2018 £’000 |
Long-term security deposits | | | 2,532 | | | 2,532 |
Prepayments and accrued income | | | 1,858 | | | — |
| | | 4,390 | | | 2,532 |
| | | 2019 £’000 | | | 2018 £’000 | |
Trade receivables | | | 1,471 | | | 4,374 |
Other receivables | | | 3,667 | | | 1,631 |
Interest receivable | | | 28 | | | 167 |
Prepayments and accrued income | | | 4,473 | | | 7,566 |
| | | 9,639 | | | 13,738 |
| | | 2019 £’000 | | | 2018 £000’s | |
Cash at bank and in hand | | | 73,966 | | | 124,385 |
| | | 73,966 | | | 124,385 |
Issued share capital (0.01p per share) | | | Growth shares | | | Series A shares | | | Series B shares | | | Ordinary shares |
At January 1, 2018 | | | 155,246 | | | 1,699,576 | | | — | | | 2,459,363 |
New shares issued for cash | | | — | | | — | | | — | | | 10,950 |
Repurchased and cancelled | | | (36,800) | | | — | | | — | | | — |
At December 31, 2018 | | | 118,446 | | | 1,699,576 | | | — | | | 2,470,313 |
New shares issued for cash | | | — | | | — | | | 621,556 | | | 45,581 |
Repurchased and cancelled | | | (60,240) | | | — | | | — | | | — |
At December 31, 2019 | | | 58,206 | | | 1,699,576 | | | 621,556 | | | 2,515,894 |
| | | 2019 £ | | | 2018 £ | |
Allotted, called up and fully paid | | | | | ||
Ordinary shares | | | 252 | | | 247 |
Series A shares | | | 170 | | | 170 |
Series B shares | | | 62 | | | — |
Growth shares | | | 6 | | | 12 |
| | | 490 | | | 429 |
| | | £’000 | |
At January 1, 2018 | | | 223,986 |
New shares issued for cash | | | 101 |
At December 31, 2018 | | | 224,087 |
New shares issued for cash | | | 59,163 |
At December 31, 2019 | | | 283,250 |
• | managing the budgeting process; |
• | managing funding and liquidity risk; and |
• | maintaining strong investor relations. |
| | | 2019 £’000 | | | 2018 £’000 | |
Long-term convertible loan (see Note 23) | | | — | | | 18,878 |
| | | — | | | 18,878 |
| | | 2019 £’000 | | | 2018 £’000 | |
Deferred revenue | | | 47,961 | | | 68,795 |
Deferred rent | | | — | | | 1,870 |
| | | 47,961 | | | 70,665 |
| | | Total £’000 | |
At January 1, 2018 | | | 267 |
Arising during the year | | | 50 |
Utilized | | | (100) |
At December 31, 2018 | | | 217 |
Arising during the year | | | 150 |
Utilized | | | (79) |
At December 31, 2019 | | | 288 |
Current | | | 183 |
Non-current | | | 105 |
| | | 2019 £’000 | | | 2018 £000’s | |
Short-term convertible loan (see Note 23) | | | 19,157 | | | — |
| | | 19,157 | | | — |
| | | 2019 £’000 | | | 2018 £’000 | |
Trade payables | | | 15,729 | | | 6,444 |
Other taxation and social security | | | 522 | | | 640 |
Accruals | | | 13,250 | | | 12,471 |
| | | 29,501 | | | 19,555 |
| | | 2019 £’000 | | | 2018 £’000 | |
Deferred revenue | | | 28,457 | | | 29,437 |
Deferred rent | | | 65 | | | 304 |
| | | 28,522 | | | 29,741 |
| | | 2019 £’000 | | | 2018 £’000 | |
Tax payable | | | 72 | | | 139 |
| | | 72 | | | 139 |
Number of shares issuable | | | Number of share options (#) | | | Weighted average exercise price (£) |
Outstanding at January 1, 2018 | | | 227,608 | | | 54.01 |
Awards granted | | | — | | | — |
Awards exercised | | | (10,950) | | | 9.26 |
Awards forfeited | | | (67,935) | | | 53.57 |
Outstanding at December 31, 2018 | | | 148,723 | | | 57.50 |
Awards granted | | | 582,252 | | | 150.00 |
Awards exercised | | | (8,574) | | | 2.71 |
Awards forfeited | | | (6,578) | | | 103.17 |
Outstanding at December 31, 2019 | | | 715,823 | | | 132.89 |
Exercisable at December 31, 2019 | | | 125,305 | | | 53.09 |
Number of shares issuable | | | Number of growth shares | | | Weighted average hurdle rate £ |
Outstanding at January 1, 2018 | | | 155,246 | | | 170.00 |
Awards granted | | | — | | | — |
Awards exercised | | | — | | | — |
Awards forfeited | | | (36,800) | | | 170.00 |
Outstanding at December 31, 2018 | | | 118,446 | | | 170.00 |
Number of shares issuable | | | Number of growth shares | | | Weighted average hurdle rate £ |
Awards granted | | | — | | | — |
Awards exercised | | | — | | | — |
Awards forfeited | | | (60,240) | | | 170.00 |
Outstanding at December 31, 2019 | | | 58,206 | | | 170.00 |
Exercisable at December 31, 2019 | | | 14,004 | | | 170.00 |
| | | Growth Shares | | | Share options | |||||||||||||
| | | Hurdle rate £ | | | Number of options | | | Weighted average remaining contractual life | | | Exercise price £ | | | Number of options | | | Weighted average remaining contractual life | |
| | | 170.00 | | | 58,206 | | | 7.3 | | | 1.99 | | | 1,563 | | | 1.7 | |
| | | — | | | — | | | — | | | 43.37 | | | 111,319 | | | 5.2 | |
| | | — | | | — | | | — | | | 120.87 | | | 3,309 | | | 6.0 | |
| | | — | | | — | | | — | | | 150.00 | | | 599,632 | | | 9.3 | |
| | | Growth shares April 2017 | | | Share options May 2019 | | | Share options April 2017 | | | Share options 2016 | |
Share price at grant date | | | £150.00 | | | £64.00 | | | £150.00 | | | £140.00 |
Exercise price | | | — | | | £150.00 | | | £150.00 | | | £43.37 - £150.00 |
Hurdle rate | | | £170.00 | | | — | | | — | | | — |
Expected volatility | | | 65% | | | 67% | | | 65% | | | 60% |
Expected life (years) | | | 2.7 yrs | | | 1.9 yrs - 3 yrs | | | 5 yrs | | | 5 yrs |
Risk free rate | | | 0.15% | | | 0.69% - 0.71% | | | 0.42% | | | 0.62% - 1.41% |
Fair value | | | £58.55 | | | £11.95 | | | £80.63 | | | £77.16 - £107.94 |
At December 31, 2019 | | | Carrying amount £’000 | | | Contractual cash flows £’000 | | | One year or less £’000 |
Financial assets | | | | | | | |||
Trade receivables | | | 1,471 | | | 1,471 | | | 1,471 |
Interest receivable | | | 28 | | | 28 | | | 28 |
Prepayments and accrued income | | | 2,282 | | | 2,282 | | | 424 |
Long-term security deposits | | | 2,532 | | | 2,532 | | | — |
Cash and cash equivalents | | | 73,966 | | | 73,966 | | | 73,966 |
Total financial assets | | | 80,279 | | | 80,279 | | | 75,889 |
Financial liabilities | | | | | | | |||
Trade payables | | | 15,579 | | | 15,579 | | | 15,579 |
Interest-bearing loans and borrowings (see Note 21) | | | 19,157 | | | 19,426 | | | 19,157 |
Derivative liability | | | 5,127 | | | — | | | 5,127 |
Total financial liabilities | | | 39,863 | | | 35,005 | | | 39,863 |
At December 31, 2018 | | | Carrying amount £’000 | | | Contractual cash flows £’000 | | | One year or less £’000 |
Financial assets | | | | | | | |||
Trade receivables | | | 4,374 | | | 4,374 | | | 4,374 |
Interest receivable | | | 167 | | | 167 | | | 167 |
Prepayments and accrued income | | | 2,660 | | | 2,660 | | | 2,660 |
Long-term security deposits | | | 2,532 | | | 2,532 | | | — |
Cash and cash equivalents | | | 124,385 | | | 124,385 | | | 124,385 |
Total financial assets | | | 134,118 | | | 134,118 | | | 131,586 |
Financial liabilities | | | | | | | |||
Trade payables | | | 6,444 | | | 6,444 | | | 6,444 |
Interest-bearing loans and borrowings (Note 19) | | | 18,878 | | | 20,096 | | | — |
Total financial liabilities | | | 25,322 | | | 26,540 | | | 6,444 |
| | | 2019 Carrying amount £’000 | | | 2018 Carrying amount £’000 | |
Cash and cash equivalents | | | 73,966 | | | 124,385 |
| | | 73,966 | | | 124,385 |
| | | 2019 Carrying amount £’000 | | | 2018 Carrying amount £’000 | |
Financial assets at amortized cost: | | | | | ||
Interest receivable | | | 15 | | | 137 |
Prepayments and accrued income | | | 1,858 | | | 2,405 |
Cash and cash equivalents | | | 12,518 | | | 86,251 |
| | | 14,391 | | | 88,793 |
| | | 2019 Carrying amount £’000 | | | 2018 Carrying amount £’000 | |
Financial liabilities at amortized cost: | | | | | ||
Trade payables | | | 4,374 | | | 2,637 |
Interest-bearing loans and borrowings (see Note 21) | | | 19,157 | | | 18,878 |
| | | 23,531 | | | 21,515 |
| | | 2019 | | | 2018 | |||||||
| | | Carrying amount £’000 | | | Fair value £’000 | | | Carrying amount £’000 | | | Fair value £’000 | |
Financial assets at amortized cost: | | | | | | | | | ||||
Trade receivables | | | 1,471 | | | 1,471 | | | 4,374 | | | 4,374 |
Interest receivable | | | 28 | | | 28 | | | 167 | | | 167 |
Prepayments and accrued income | | | 2,282 | | | 2,282 | | | 2,660 | | | 2,660 |
Long-term security deposits | | | 2,532 | | | 2,532 | | | 2,532 | | | 2,532 |
Embedded derivative asset | | | 266 | | | 266 | | | 719 | | | 719 |
Cash and cash equivalents | | | 73,966 | | | 73,966 | | | 124,385 | | | 124,385 |
Total financial assets at amortized cost | | | 80,545 | | | 80,545 | | | 134,837 | | | 134,837 |
| | | 2019 | | | 2018 | |||||||
| | | Carrying amount £’000 | | | Fair value £’000 | | | Carrying amount £’000 | | | Fair value £’000 | |
Financial liabilities at amortized cost | | | | | | | | | ||||
Trade payables | | | 15,579 | | | 15,579 | | | 6,444 | | | 6,444 |
Interest-bearing loans and borrowings (Note 21) | | | 19,157 | | | 19,157 | | | 18,878 | | | 18,878 |
Derivative liability | | | 5,127 | | | 5,127 | | | — | | | — |
Total financial liabilities | | | 39,863 | | | 39,863 | | | 25,322 | | | 25,322 |
| | | Interest rate % | | | Maturity date £000 | | | 2019 £000 | | | 2018 £000 | |
Gates Foundation convertible loan | | | Variable | | | September 12, 2020 | | | 19,157 | | | 18,878 |
| | | At January 1, 2019 £’000 | | | Cash flows £’000 | | | Foreign exchange movement £’000 | | | Net finance (income) / costs £’000 | | | Leases £’000 | | | At December 31, 2019 £’000 | |
Interest-bearing loans and borrowings | | | 18,878 | | | — | | | (563) | | | 842 | | | — | | | 19,157 |
Derivative liability | | | — | | | — | | | — | | | 5,127 | | | — | | | 5,127 |
Lease liabilities | | | 46,555 | | | (4,036) | | | 9 | | | 2,938 | | | (5,216) | | | 40,250 |
Total liabilities from financing activities | | | 65,433 | | | (4,036) | | | (554) | | | 8,907 | | | (5,216) | | | 64,534 |
| | | At January 1, 2018 £’000 | | | Foreign exchange movement £’000 | | | Interest expense £’000 | | | At December 31, 2018 £’000 | |
Interest-bearing loans and borrowings | | | 16,940 | | | 1,096 | | | 842 | | | 18,878 |
Total liabilities from financing activities | | | 16,940 | | | 1,096 | | | 842 | | | 18,878 |
As at December 31, 2019 | | | Less than 1 year | | | 1-3 years | | | 3-5 years | | | More than 5 years | | | Total |
Lease liabilities – existing | | | 4,469 | | | 8,958 | | | 7,876 | | | 45,288 | | | 66,591 |
Lease liabilities – contingent | | | 68 | | | 1,604 | | | 2,685 | | | 2,688 | | | 7,045 |
Manufacturing | | | 3,669 | | | 642 | | | — | | | — | | | 4,311 |
Capital commitments | | | 1,460 | | | — | | | — | | | — | | | 1,460 |
Total contractual obligations | | | 9,666 | | | 11,204 | | | 10,561 | | | 47,976 | | | 79,407 |
As at December 31, 2018 | | | Less than 1 year | | | 1-3 years | | | 3-5 years | | | More than 5 years | | | Total |
Operating lease payables | | | 4,329 | | | 8,467 | | | 8,099 | | | 60,691 | | | 81,586 |
Manufacturing | | | 10,544 | | | 55 | | | — | | | — | | | 10,599 |
Capital commitments | | | 347 | | | — | | | — | | | — | | | 347 |
Total contractual obligations | | | 15,220 | | | 8,522 | | | 8,099 | | | 60,691 | | | 92,532 |
| | | 2019 | | | 2018 | |||||||
| | | Sales to related party £000’s | | | Purchases from related party £000’s | | | Sales to related party £000’s | | | Purchases from related party £000’s | |
Adaptimmune Limited | | | — | | | — | | | 69 | | | — |
Aigenpulse Limited | | | — | | | 500 | | | — | | | 729 |
Malin Life Sciences Holdings Limited | | | — | | | — | | | — | | | 2 |
Oxford Nanosystems Limited | | | — | | | — | | | 2 | | | — |
Oxford Innovation Ltd | | | — | | | 30 | | | — | | | 13 |
| | | — | | | 530 | | | 71 | | | 744 | |
| | | 2019 | | | 2018 | |||||||
| | | Receivables outstanding from related party £000’s | | | Payables outstanding to related party £000’s | | | Receivables outstanding from related party £000’s | | | Payables outstanding to related party £000’s | |
Aigenpulse Limited | | | — | | | — | | | — | | | 345 |
Adaptimmune Limited | | | — | | | — | | | 11 | | | — |
Oxford Nanosystems Limited | | | — | | | — | | | 2 | | | — |
Oxford Innovation Ltd | | | — | | | — | | | — | | | 1 |
| | | — | | | — | | | 13 | | | 346 | |
| | | 2019 £000’s | | | 2018 £000’s | |
Short-term employee benefits | | | 6,502 | | | 4,435 |
Share-based payments | | | 3,667 | | | 270 |
| | | 10,169 | | | 4,705 |
| | | Notes | | | 2020 £’000 | | | 2019 £’000 | |
Revenue | | | 2 | | | 22,694 | | | 20,027 |
Total revenue | | | | | 22,694 | | | 20,027 | |
| | | | | | | ||||
Other operating income | | | | | 408 | | | 420 | |
Research and development costs | | | | | (57,566) | | | (75,415) | |
Administrative expenses | | | | | (31,569) | | | (35,611) | |
Operating loss | | | | | (66,033) | | | (90,579) | |
| | | | | | | ||||
Finance income | | | 3 | | | 1,972 | | | 1,134 |
Finance costs | | | 4 | | | (2,272) | | | (6,532) |
Non-operating expense | | | | | (300) | | | (5,398) | |
| | | | | | | ||||
Loss before taxation | | | | | (66,333) | | | (95,977) | |
Income tax credit | | | 5 | | | 11,120 | | | 18,011 |
Loss for the period | | | | | (55,213) | | | (77,966) | |
| | | | | | | ||||
Other comprehensive income | | | | | | | |||
Other comprehensive income that are or may be reclassified to profit or loss in subsequent periods (net of tax): | | | | | | | |||
Exchange differences on translation of foreign operations | | | | | 338 | | | 82 | |
Total other comprehensive income for the period, net of tax | | | | | 338 | | | 82 | |
| | | | | | | ||||
Total comprehensive loss for the period, net of tax | | | | | (54,875) | | | (77,884) | |
Basic and diluted loss per share | | | 6 | | | (0.01) | | | (0.02) |
| | | Notes | | | September 30, 2020 £’000 | | | December 31, 2019 £’000 | |
Non-current assets | | | | | | | |||
Property, plant and equipment | | | 7 | | | 45,068 | | | 54,880 |
Investment in sub-lease | | | | | 384 | | | 591 | |
Other non-current financial assets | | | 8 | | | 7,003 | | | 4,390 |
Deferred tax asset | | | | | 1,539 | | | 1,507 | |
Total non-current assets | | | | | 53,994 | | | 61,368 | |
Current assets | | | | | | | |||
Trade and other receivables | | | 9 | | | 7,479 | | | 9,639 |
Tax receivable | | | | | 12,679 | | | 40,410 | |
Embedded derivative assets | | | 10 | | | — | | | 266 |
Cash and cash equivalents | | | 11 | | | 56,687 | | | 73,966 |
Total current assets | | | | | 76,845 | | | 124,281 | |
Total assets | | | | | 130,839 | | | 185,649 | |
Equity | | | | | | | |||
Share capital | | | 12 | | | 1 | | | — |
Share premium | | | 12 | | | 330,390 | | | 283,250 |
Foreign currency translation reserve | | | 12 | | | 306 | | | (32) |
Share-based payment reserve | | | 12, 13 | | | 15,840 | | | 10,659 |
Accumulated deficit | | | | | (330,989) | | | (279,106) | |
Total equity | | | | | 15,548 | | | 14,771 | |
| | | | | | | ||||
Non-current liabilities | | | | | | | |||
Deferred liabilities | | | | | 35,682 | | | 47,961 | |
Lease liabilities | | | 14 | | | 31,861 | | | 38,299 |
Provisions | | | | | 238 | | | 105 | |
Total non-current liabilities | | | | | 67,781 | | | 86,365 | |
| | | | | | | ||||
Current liabilities | | | | | | | |||
Interest-bearing loans and borrowings | | | 10 | | | — | | | 19,157 |
Trade and other payables | | | 15 | | | 23,280 | | | 29,501 |
Deferred liabilities | | | | | 22,132 | | | 28,522 | |
Tax payable | | | | | — | | | 72 | |
Lease liabilities | | | 14 | | | 2,098 | | | 1,951 |
Derivative liabilities | | | | | — | | | 5,127 | |
Provisions | | | | | — | | | 183 | |
Total current liabilities | | | | | 47,510 | | | 84,513 | |
Total liabilities | | | | | 115,291 | | | 170,878 | |
Total equity and liabilities | | | | | 130,839 | | | 185,649 |
| | | Notes | | | Share capital £’000 | | | Share premium £’000 | | | Foreign currency translation reserve £’000 | | | Share- based payment reserve £’000 | | | Accumulated deficit £’000 | | | Total equity £’000 | |
At January 1, 2019 | | | | | — | | | 224,087 | | | 67 | | | 7,603 | | | (175,175) | | | 56,582 | |
Loss for the period | | | | | — | | | — | | | — | | | — | | | (77,966) | | | (77,966) | |
Other comprehensive income | | | | | — | | | — | | | 82 | | | — | | | — | | | 82 | |
Total comprehensive loss for the period | | | | | — | | | — | | | 82 | | | — | | | (77,966) | | | (77,884) | |
Issue of share capital | | | 12 | | | — | | | 59,162 | | | — | | | — | | | — | | | 59,162 |
Equity-settled share-based payment transactions | | | 12, 13 | | | — | | | — | | | — | | | 2,501 | | | — | | | 2,501 |
At September 30, 2019 | | | | | — | | | 283,249 | | | 149 | | | 10,104 | | | (253,141) | | | 40,361 | |
| | | | | | | | | | | | | | | ||||||||
As at January 1, 2020 | | | | | | | 283,250 | | | (32) | | | 10,659 | | | (279,106) | | | 14,771 | ||
Loss for the period | | | | | — | | | — | | | — | | | — | | | (55,213) | | | (55,213) | |
Other comprehensive income | | | | | — | | | — | | | 338 | | | — | | | — | | | 338 | |
Total comprehensive loss for the period | | | | | — | | | — | | | 338 | | | — | | | (55,213) | | | (54,875) | |
Conversion of interest-bearing loan | | | 10 | | | — | | | — | | | — | | | — | | | (510) | | | (510) |
Derecognition of derivative liability | | | 3 | | | — | | | — | | | — | | | — | | | 3,840 | | | 3,840 |
Issue of share capital | | | 12 | | | 1 | | | 47,140 | | | — | | | — | | | — | | | 47,141 |
Equity-settled share-based payment transactions | | | 12, 13 | | | — | | | — | | | — | | | 5,181 | | | — | | | 5,181 |
At September 30, 2020 | | | | | 1 | | | 330,390 | | | 306 | | | 15,840 | | | (330,989) | | | 15,548 |
| | | 2020 £’000 | | | 2019 £’000 | |
Cash flows from operating activities | | | | | ||
Loss for the period | | | (55,213) | | | (77,966) |
Adjustments for: | | | | | ||
Depreciation of property, plant and equipment | | | 6,652 | | | 6,691 |
Amortization of intangible assets | | | — | | | 210 |
Write-off of intangible assets | | | — | | | 306 |
Loss on disposal of property, plant and equipment | | | (148) | | | (26) |
Net finance costs | | | 300 | | | 5,398 |
Movement in provisions and other charges | | | (50) | | | 2,392 |
Foreign exchange translation differences | | | 326 | | | 608 |
Equity settled share-based payment expenses | | | 5,181 | | | 2,501 |
Taxation charge | | | (11,120) | | | (18,011) |
Working capital adjustments: | | | | | ||
(Increase)/decrease in trade and other receivables | | | (612) | | | (857) |
(Decrease)/increase in trade and other payables | | | (6,224) | | | 5,784 |
Decrease in deferred liabilities | | | (18,670) | | | (17,747) |
Cash used in operations | | | (79,578) | | | (90,717) |
Bank interest received on cash and cash equivalents | | | 676 | | | 1,202 |
Net taxation received | | | 38,904 | | | 13,825 |
Net cash used in operating activities | | | (39,998) | | | (75,690) |
Cash flows from investing activities | | | | | ||
Proceeds from sale of property, plant and equipment | | | 52 | | | 82 |
Purchase of property, plant and equipment | | | (2,727) | | | (2,449) |
Lease capital contribution | | | 1,088 | | | |
Purchase of intangible assets | | | — | | | (198) |
Proceeds from sub-leases | | | 241 | | | 22 |
Net cash flows used in investing activities | | | (1,346) | | | (2,543) |
Cash flows from financing activities | | | | | ||
Proceeds from exercise of share options | | | 45 | | | — |
Gross proceeds from issue of share capital | | | 27,288 | | | 59,901 |
Costs from issue of share capital | | | (58) | | | (738) |
Repayment of lease liabilities | | | (3,297) | | | (2,991) |
Net cash flows from financing activities | | | 23,978 | | | 56,172 |
Increase/(decrease) in net cash and cash equivalents | | | (17,366) | | | (22,061) |
Net foreign exchange difference on cash held | | | 87 | | | 74 |
Cash and cash equivalents at beginning of the year | | | 73,966 | | | 124,385 |
Cash and cash equivalents at end of the year | | | 56,687 | | | 102,398 |
• | the Group has key worker status which allows continuity of providing services throughout a prolonged lockdown period; |
• | the Group has a track record of meeting expectations under its collaboration agreements and meeting expected milestones within the contracted timeframe; |
• | the Group’s history of being able to access equity and loan financing as and when needed; and |
• | the Group’s ability and history to control capital expenditure costs and lower other operational spend, as necessary. |
• | whether achievement of a development milestone is highly susceptible to factors outside the entity’s influence, such as milestones involving the judgment or actions of third parties, including regulatory bodies or the customer; |
• | whether the uncertainty about the achievement of the milestone is not expected to be resolved for a long period of time; |
• | whether the Company can reasonably predict that a milestone will be achieved based on previous experience; and. |
• | the complexity and inherent uncertainty underlying the achievement of the milestone. |
• | adjustments arising from a change in the estimate of when the performance obligation will have been completed. |
• | adjustment to revenue that affects deferred revenue; |
• | a change in the estimate of the transaction price due to changes in the assessment of whether variable consideration is constrained because it is not considered probable of being received; and |
• | the recognition of revenue. |
• | past history and experience with similar contracts. |
• | unexpected fluctuations in planned spend. |
• | changes to project timelines. |
| | | For the nine months ended September 30, 2020 £’000 | | | For the nine months ended September 30, 2019 £’000 | |
GlaxoSmithKline | | | 4,344 | | | 3,796 |
Eli Lilly | | | 3,522 | | | 1,886 |
Genentech | | | 14,828 | | | 14,345 |
| | | 22,694 | | | 20,027 | |
| | | | | |||
United Kingdom | | | 4,344 | | | 3,796 |
United States | | | 18,350 | | | 16,231 |
| | | 22,694 | | | 20,027 |
| | | At January 1, 2020 £’000 | | | Additions £’000 | | | Deductions £’000 | | | At September 30, 2020 £’000 | |
Trade receivables: | | | | | | | | | ||||
Collaboration agreement trade receivables | | | 1,186 | | | 3,076 | | | (4,262) | | | — |
Total receivables | | | 1,186 | | | 3,076 | | | (4,262) | | | — |
Contract assets: | | | | | | | | | ||||
Contract assets | | | 424 | | | 920 | | | (424) | | | 920 |
Total contract assets | | | 424 | | | 920 | | | (424) | | | 920 |
Contract liabilities: | | | | | | | | | ||||
Deferred revenue | | | 76,418 | | | — | | | (18,698) | | | 57,720 |
Total contract liabilities | | | 76,418 | | | — | | | (18,698) | | | 57,720 |
| | | At January 1, 2019 £’000 | | | Additions £’000 | | | Deductions £’000 | | | At December 31, 2019 £’000 | |
Trade receivables: | | | | | | | | | ||||
Collaboration agreement trade receivables | | | 3,600 | | | 3,431 | | | (5,845) | | | 1,186 |
Total receivables | | | 3,600 | | | 3,431 | | | (5,845) | | | 1,186 |
Contract assets: | | | | | | | | | ||||
Contract assets | | | — | | | 424 | | | — | | | 424 |
Total contract assets | | | — | | | 424 | | | — | | | 424 |
Contract liabilities: | | | | | | | | | ||||
Deferred revenue | | | 98,232 | | | — | | | (21,814) | | | 76,418 |
Total contract liabilities | | | 98,232 | | | — | | | (21,814) | | | 76,418 |
| | | At September 30, 2020 £’000 | | | At December 31, 2019 £’000 | |
Current deferred revenue: | | | | | ||
GlaxoSmithKline | | | 3,253 | | | 3,895 |
Eli Lilly | | | 1,696 | | | 7,151 |
Genentech | | | 17,089 | | | 17,411 |
Current deferred revenue | | | 22,038 | | | 28,457 |
Non-current deferred revenue: | | | | | ||
GlaxoSmithKline | | | 2,247 | | | 3,603 |
Eli Lilly | | | 5,665 | | | 3,732 |
Genentech | | | 27,770 | | | 40,626 |
Non-current deferred revenue | | | 35,682 | | | 47,961 |
Total deferred revenue | | | 57,720 | | | 76,418 |
| | | For the nine months ended September 30, 2020 £’000 | | | For the nine months ended September 30, 2019 £’000 | |
Interest on cash and cash equivalents | | | 660 | | | 1,107 |
Lease interest income | | | 25 | | | 27 |
Gain from change in fair value of derivative liability | | | 1,287 | | | — |
| | | 1,972 | | | 1,134 |
| | | For the nine months ended September 30, 2020 £’000 | | | For the nine months ended September 30, 2019 £’000 | |
Interest on lease liabilities | | | 1,847 | | | 2,283 |
Interest expenses on financial liabilities measured at amortized cost | | | 159 | | | 694 |
Loss from change in fair value of embedded derivative asset | | | 266 | | | 438 |
Loss from change in fair value of derivative liability | | | — | | | 3,117 |
| | | 2,272 | | | 6,532 |
| | | For the nine months ended September 30, 2020 £’000 | | | For the nine months ended September 30, 2019 £’000 | |
Loss for the period | | | (55,213) | | | (77,966) |
Basic and diluted weighted average number of shares | | | 5,458,712 | | | 4,315,976 |
Basic and diluted loss per share | | | (0.01) | | | (0.02) |
| | | September 30, 2020 £’000 | | | December 31, 2019 £000 | |
Long-term security deposits | | | 2,532 | | | 2,532 |
Prepayments and accrued income | | | 4,471 | | | 1,858 |
| | | 7,003 | | | 4,390 |
| | | September 30, 2020 £’000 | | | December 31, 2019 £000 | |
Trade receivables | | | 136 | | | 1,471 |
Other receivables | | | 1,861 | | | 3,667 |
Interest receivables | | | — | | | 28 |
Prepayments and accrued income | | | 5,482 | | | 4,473 |
| | | 7,479 | | | 9,639 |
| | | September 30, 2020 £’000 | | | December 31, 2019 £000 | |
Cash at bank and in hand | | | 56,687 | | | 73,966 |
| | | 56,687 | | | 73,966 |
Issued share capital (0.01p per share) | | | Growth shares | | | Series A shares | | | Series B shares | | | Ordinary shares |
At January 1, 2020 | | | 58,206 | | | 1,699,576 | | | 621,556 | | | 2,515,894 |
New shares issued for cash | | | 34,260 | | | — | | | 323,450 | | | 35,730 |
New shares issued for non-cash consideration | | | — | | | — | | | 203,697 | | | — |
Repurchased and cancelled | | | (29,916) | | | — | | | — | | | — |
At September 30, 2020 | | | 62,550 | | | 1,699,576 | | | 1,148,703 | | | 2,551,624 |
| | | 2020 £ | | | 2019 £ | |
Allotted, called up and fully paid | | | | | ||
Ordinary shares | | | 255 | | | 252 |
Series A shares | | | 170 | | | 170 |
Series B shares | | | 115 | | | 62 |
Growth shares | | | 6 | | | 6 |
| | | 546 | | | 490 |
| | | £’000 | |
At January 1, 2020 | | | 283,250 |
New shares issued for cash | | | 27,275 |
New shares issued for non-cash consideration | | | 19,865 |
At September 30, 2020 | | | 330,390 |
Number of shares issuable | | | Number of share options (#) | | | Weighted average exercise price (£) |
Outstanding at January 1, 2020 | | | 715,823 | | | 132.89 |
Awards granted | | | 144,370 | | | 64.00 |
Awards exercised | | | (2,529) | | | 17.80 |
Awards forfeited | | | (24,760) | | | 67.43 |
Outstanding at September 30, 2020 | | | 832,904 | | | 63.23 |
Exercisable at September 30, 2020 | | | 220,519 | | | 59.44 |
Number of shares issuable | | | Number of growth shares | | | Weighted average hurdle rate £ |
Outstanding at January 1, 2020 | | | 58,206 | | | 170.00 |
Awards granted | | | 34,260 | | | 64.00 |
Awards exercised | | | — | | | — |
Awards forfeited | | | (29,916) | | | 170.00 |
Outstanding at September 30, 2020 | | | 62,550 | | | 137.36 |
Exercisable at September 30, 2020 | | | 36,761 | | | 156.78 |
| | | Growth Shares | | | Share options | |||||||||||||
| | | Hurdle rate £ | | | Number of options | | | Weighted average remaining contractual life | | | Exercise price £ | | | Number of options | | | Weighted average remaining contractual life | |
| | | 170.00 | | | 43,290 | | | 7.6 | | | 43.37 | | | 92,544 | | | 4.4 | |
| | | 64.00 | | | 19,260 | | | 9.6 | | | 120.87 | | | 3,309 | | | 5.3 | |
| | | | | | | | | 150.00 | | | 12,585 | | | 6.5 | ||||
| | | | | | | | | 64.00 | | | 724,466 | | | 8.5 | ||||
| | | Growth shares | | | Share options | |
Share price at grant date | | | £64.00 | | | £64.00 |
Exercise price | | | — | | | £64.00 |
Hurdle rate | | | £64.00 | | | — |
Expected volatility | | | 91% - 102% | | | 78% - 93% |
Expected life (years) | | | 1 yrs | | | 1.6 - 3 yrs |
Risk-free rate | | | (0.02%) -0.03% | | | (0.03%) - 0.13% |
Fair value | | | £2.12 - £7.71 | | | £28.44 - £34.30 |
| | | September 30, 2020 £’000 | | | December 31, 2019 £000 | |
Current | | | 2,098 | | | 1,951 |
Non-current | | | 31,861 | | | 38,299 |
Total lease liabilities | | | 33,959 | | | 40,250 |
| | | September 30, 2020 £’000 | | | December 31, 2019 £000 | |
Trade payables | | | 5,829 | | | 15,729 |
Other taxation and social security | | | 607 | | | 522 |
Accruals | | | 16,844 | | | 13,250 |
| | | 23,280 | | | 29,501 |
£000s | | | Less than 1 year | | | 1-3 years | | | 3-5 Years | | | More than 5 years | | | Total |
Lease liabilities – existing | | | 4,275 | | | 7,910 | | | 6,548 | | | 35,657 | | | 54,390 |
Lease liabilities – contingent | | | — | | | 2,254 | | | 2,471 | | | 1,841 | | | 6,566 |
Manufacturing | | | 2,021 | | | 471 | | | — | | | — | | | 2,492 |
Capital commitments | | | 2,134 | | | — | | | — | | | — | | | 2,134 |
Total contractual obligations | | | 8,430 | | | 10,635 | | | 9,019 | | | 37,498 | | | 65,582 |
| | | For the nine months ended September 30, 2020 | | | For the year ended December 31, 2019 | |||||||
| | | Sales to related party £000 | | | Purchases from related party £000 | | | Sales to related party £000 | | | Purchases from related party £000 | |
Aigenpulse Limited | | | — | | | — | | | — | | | 500 |
Oxford Innovation Limited | | | — | | | — | | | — | | | 30 |
| | | — | | | — | | | — | | | 530 | |
| | | For the nine months ended September 30, 2020 £000 | | | For the nine months ended September 30, 2019 £000 | |
Short-term employee benefits | | | 2,084 | | | 5,827 |
Share-based payments | | | 3,416 | | | 2,874 |
| | | 5,500 | | | 8,701 |
Goldman Sachs & Co. LLC | | | J.P. Morgan | | | Jefferies |
Item 6. | Indemnification of Directors and Officers. |
Item 7. | Recent Sales of Unregistered Securities. |
• | On April 19, 2017, Immunocore Limited issued 155,246 G1 shares to Immunocore Nominees Limited at a purchase price of £0.0001 per share for an aggregate consideration of £15.52. |
• | On April 19, 2017, Immunocore Limited issued 10,000 ordinary shares to an option holder upon the exercise of share options at an exercise price of £43.37 per share for an aggregate consideration of £433,700.00. |
• | In August 2018, September 2018, October 2018, November 2018 and December 2018, Immunocore Limited issued an aggregate of 10,960 ordinary shares to Immunocore Nominees Limited at purchase prices ranging from £0.74 to £150 per share for an aggregate consideration of £101,409.74. |
• | In January 2019, February 2019, and March 2019, Immunocore Limited issued an aggregate of 4,267 ordinary shares to Immunocore Nominees Limited at purchase prices ranging from £0.74 to £1.99 per share for an aggregate consideration of £4,020.08. |
• | In April 2019 and June 2019, Immunocore Limited issued an aggregate of 3,043 ordinary shares to Immunocore Nominees Limited at purchase prices ranging from £0.74 to £1.99 per share for an aggregate consideration of £5,373.10. |
• | In July 2019 and September 2019, Immunocore Limited issued an aggregate of 345 ordinary shares to Immunocore Nominees Limited at purchase prices ranging from £1.99 to £43.37 per share for an aggregate consideration of £11,155.69. |
• | On November 4, 2019, Immunocore Limited issued 919 ordinary shares to Immunocore Nominees Limited at a purchase price of £1.99 per share for aggregate consideration of £1,828.81. |
• | On December 19, 2019, Immunocore Limited issued an aggregate of 37,007 ordinary shares to 30 accredited investors and insiders at a purchase price of £0.0001 per share for aggregate consideration of £3.70. |
• | On January 9, 2020, Immunocore Limited issued 360 ordinary shares to Immunocore Nominees Limited at a purchase price of £1.99 per share for aggregate consideration of £714.60. |
• | On February 17, 2020, Immunocore Limited issued 184 ordinary shares to Immunocore Nominees Limited at a purchase price of £1.99 per share for aggregate consideration of £366.16. |
• | On February 24, 2020, Immunocore Limited issued 25 ordinary shares to Immunocore Nominees Limited at a purchase price of £43.37 per share for aggregate consideration of £1,084.25. |
• | On March 2, 2020, Immunocore Limited issued an aggregate of 33,201 ordinary shares to 30 insiders and accredited investors at a purchase price of £0.0001 per share for an aggregate consideration of £3.32. |
• | On March 9, 2020, Immunocore Limited issued 500 ordinary shares to Immunocore Nominees Limited at a purchase price of £1.99 per share for an aggregate consideration of £995.00. |
• | On March 19, 2020, Immunocore Limited issued 289 ordinary shares to Immunocore Nominees Limited at a purchase price of £1.99 per share for aggregate consideration of £575.11. |
• | In June 2020, Immunocore Limited issued 941 ordinary shares to Immunocore Nominees Limited at a purchase price of £43.37 per share for aggregate consideration of £40,811.17. |
• | On September 3, 2020, Immunocore Limited issued 230 ordinary shares to Immunocore Nominees Limited at a purchase price of £1.99 per share for aggregate consideration of £457.70. |
• | On October 19, 2020, Immunocore Limited issued 247 ordinary shares to Immunocore Nominees Limited at a purchase price of £64 per share for aggregate consideration of £15,808. |
• | In August 2019, Immunocore Limited issued an aggregate of 621,556 series B preferred shares to 5 insiders and accredited investors at a purchase price of £96.19 per share for an aggregate consideration of £59,787,471.64. |
• | In March 2020, Immunocore Limited issued an aggregate of 527,147 series B preferred shares to 10 insiders and accredited investors at purchase prices ranging from £73.91 to £96.19 per share for an aggregate consideration of £49,747,271.89. |
• | In December 2020, Immunocore Limited issued an aggregate of 823,719 series C preferred shares to four insiders and accredited investors and third party accredited investors at a purchase price of $91.05 per share for an aggregate consideration of $74,999,614.95. At the same time, Immunocore Limited issued 127,893 ordinary shares to insiders by way of capitalization of our undistributable reserves pursuant to their pre-existing anti-dilution rights. |
Grant Date | | | Number of Share Options | | | Exercise Price per Share |
May 13, 2019 | | | 582,252 | | | £150.00 |
April 30, 2020 | | | 138,620 | | | £64.00 |
May 4, 2020 | | | 450 | | | £64.00 |
June 3, 2020 | | | 5,100 | | | £64.00 |
June 10, 2020 | | | 200 | | | £64.00 |
October 30, 2020 | | | 51,519 | | | £64.00 |
November 10, 2020 | | | 22,700 | | | £64.00 |
November 16, 2020 | | | 5,947 | | | £64.00 |
Grant Date | | | Number of G Shares Granted |
April 30, 2020 | | | 32,260 |
June 10, 2020 | | | 2,000 |
Item 8. | Exhibits and Financial Statement Schedules |
Exhibit Number | | | Description of Exhibit |
| | | Form of Underwriting Agreement. | |
| | | Form of Articles of Association of Immunocore Holdings Limited (to be re-registered as a public limited company and re-named Immunocore Holdings plc). | |
| | | Form of Deposit Agreement. | |
| | | Form of American Depositary Receipt (included in Exhibit 4.1). | |
| | | Form of Subscription Agreement between the Registrant and the Bill & Melinda Gates Foundation, dated February , 2021. | |
| | | Opinion of Cooley (UK) LLP. | |
| | | Form of Deed of Indemnity between the Registrant and each of its directors. | |
| | | Form of Deed of Indemnity between the Registrant and each of its executive officers. | |
| | | Form of Immunocore Holdings plc 2021 Equity Incentive Plan. | |
10.4# | | | Non-Employee Sub Plan to the Immunocore Holdings plc 2021 Equity Incentive Plan (included in Exhibit 10.3). |
| | | Research Collaboration and License Agreement, dated as of June 14, 2013, by and among the Registrant, Genentech, Inc. and F. Hoffman-La Roche Ltd, as amended on September 27, 2016. | |
| | | Collaboration and License Agreement, dated as of June 29, 2013, between the Registrant and GlaxoSmithKline Intellectual Property Development Ltd. | |
| | | Development and License Agreement, dated as of July 11, 2014, between the Registrant and Eli Lilly and Company, as amended on December 21, 2016, September 20, 2017 and December 19, 2018. | |
| | | License Agreement, dated as of September 27, 2016, between the Registrant and Genentech, Inc. | |
| | | License and Collaboration Agreement, dated as of November 15, 2018, by and among the Registrant, Genentech, Inc. and F. Hoffman-La Roche Ltd. | |
| | | Convertible Loan Note Purchase Agreement, dated as of September 13, 2017, between the Registrant and the Bill & Melinda Gates Foundation. | |
| | | Amended and Restated Global Access Commitments Agreement, dated as of March 2, 2020, between the Registrant and the Bill & Melinda Gates Foundation. | |
| | | Form of First Amendment to the Amended and Restated Global Access Commitments Agreement, dated as of February ,2021, between the Registrant and the Bill & Melinda Gates Foundation. | |
| | | Lease, dated as of March 28, 2017, between the Registrant and MEPC MILTON PARK NO. 1 LIMITED and MEPC MILTON PARK NO. 2 LIMITED, on behalf of MEPC Milton LP. | |
| | | Lease, dated as of December 28, 2017, between the Registrant and MEPC MILTON PARK NO. 1 LIMITED and MEPC MILTON PARK NO. 2 LIMITED, on behalf of MEPC Milton LP. | |
| | | Lease, dated as of March 28, 2017, between the Registrant and MEPC MILTON PARK NO. 1 LIMITED and MEPC MILTON PARK NO. 2 LIMITED, on behalf of MEPC Milton LP. | |
| | | Assignment and Exclusive License, dated as of January 28, 2015, between the Registrant and Adaptimmune Limited. | |
| | | Loan and Security Agreement, dated as of November 6, 2020, among the Registrant, Oxford Finance Luxembourg S.à r.l., and the lenders listed on Schedule 1.1 thereof. | |
| | | Employment Agreement between the Registrant and Bahija Jallal, Ph.D, dated January 29, 2021. | |
| | | Form of Deed of Termination of Convertible Loan Note Purchase Agreement, dated as of February , 2021, between the Registrant and the Bill & Melinda Gates Foundation. | |
| | | Subsidiaries of the Registrant. | |
| | | Consent of KPMG LLP, the Registrant’s independent registered public accounting firm (Immunocore Holdings Limited). | |
| | | Consent of KPMG LLP, the Registrant’s independent registered public accounting firm (Immunocore Limited). | |
23.3 | | | Consent of Cooley (UK) LLP (included in Exhibit 5.1). |
| | | Power of Attorney (included on signature page to registration statement filed January 15, 2021). | |
| | | Power of Attorney of Roy S. Herbst, M.D., Ph.D. | |
| | | Request for Waiver from Requirements of Form 20-F, Item 8.A.4, dated February 1, 2021. |
† | Certain portions of this exhibit (indicated by asterisks) have been redacted in accordance with Regulation S-K, Item 601(b)(10). |
# | Indicates a management contract or any compensatory plan, contract or arrangement. |
* | Previously filed. |
Item 9. | Undertakings. |
(1) | For purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this Registration Statement in reliance upon Rule 430A and contained in a form of prospectus filed by the Registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this Registration Statement as of the time it was declared effective. |
(2) | For the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
| | | IMMUNOCORE HOLDINGS LIMITED | |||||||
| | | | | | | ||||
| | | By: | | | /s/ Bahija Jallal, Ph.D. | ||||
| | | | | Name: | | | Bahija Jallal, Ph.D. | ||
| | | | | Title: | | | Chief Executive Officer | ||
Signature | | | Title | | | Date |
| | | | | |||
/s/ Bahija Jallal, Ph.D. | | | Chief Executive Officer and Director (Principal Executive Officer) | | | |
Bahija Jallal, Ph.D. | | | February 1, 2021 | |||
| | | | ||||
/s/ Brian Di Donato | | | Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) | | | |
Brian Di Donato | | | February 1, 2021 | |||
| | | | | |||
* | | | Chairman of the Board of Directors | | ||
Professor Sir John Bell | | | February 1, 2021 | |||
| | | | | |||
* | | | Director | | | |
Travis Coy | | | February 1, 2021 | |||
| | | | | |||
* | | | Director | | | |
Roy Herbst, M.D., Ph.D. | | | | | February 1, 2021 | |
| | | | | |||
* | | | Director | | | |
Robert Perez | | | February 1, 2021 | |||
| | | | | |||
* | | | Director | | | |
Kristine Peterson | | | February 1, 2021 | |||
| | | | | |||
* | | | Director | | | |
Professor Sir Peter Ratcliffe | | | February 1, 2021 |
*By: | | | /s/ Bahija Jalllal, Ph.D. | | | |
| | | /s/ Bahija Jalllal, Ph.D. Attorney-in-fact | | |
| | | Immunocore, LLC | ||||
| | | | | |||
| | | By: | | | /s/ Bahija Jallal, Ph.D. | |
| | | Name: | | | Bahija Jallal, Ph.D. | |
| | | Title: | | | Authorized Signatory | |